The Alzheimer's Brain Is Overloaded With Sugar-Protein Modifications. A New Study Shows What That Is Doing to Cognition.
Alzheimer's disease has been understood primarily as a disease of amyloid accumulation, but the amyloid hypothesis has produced a long record of drug trial failures. A new study published in Nature Metabolism from the University of Florida identifies a metabolic pathway that has been almost entirely overlooked: the excessive accumulation of sugar-protein modifications called glycans throughout the Alzheimer's brain, driven by increased glycan biosynthesis rather than impaired clearance.
Glycosylation is one of the most fundamental modifications the cell makes to its proteins, and the brain depends on it more heavily than almost any other organ. Glycans regulate synaptic transmission, blood-brain barrier integrity, neuroinflammatory signaling, and intracellular protein trafficking. What the new study identifies is not the absence of glycosylation but its excess, a quantitative dysregulation of a normal biological process that changes how neuronal proteins fold, traffic, and interact with their molecular partners.
Using spatial mass spectrometry imaging, the team found dramatically elevated glycan levels across both grey and white matter in human Alzheimer's brains. The elevation was widespread across multiple glycan types and progressive across disease stages: glycan abundance in grey matter increased consistently from Braak stage zero through stages five and six, with the most pronounced elevations in the most severely affected brains. More than 90 to 95 percent of the glycoproteins showing elevated glycosylation in AD were the same proteins present in normal brains, meaning what changed was not which proteins carry glycans but how heavily they are modified.
The same hyperglycosylation phenotype appeared in both an amyloid mouse model and a tau mouse model, establishing it as a common feature of neurodegeneration rather than a consequence of any single pathological protein. The glycan elevation was most pronounced in the cortex, hippocampus, and thalamus, the regions most critically affected by Alzheimer's disease, and less pronounced in the cerebellum and hindbrain, consistent with the regional vulnerability pattern of clinical disease progression.
Stable isotope tracing established that hyperglycosylation is driven by elevated glycan biosynthesis rather than impaired clearance. AD mice incorporated significantly more labeled carbon into glycans during the pulse phase than wild-type controls, while glycan turnover during the chase phase showed no significant difference between groups. The AD brain is making more glycans, not failing to clear them, and it appears to be selectively redirecting available glucose toward glycan production at the expense of two other competing biological processes that were actually reduced rather than elevated in AD mice.
The Alzheimer's brain appears to be channeling its limited glucose resources toward glycan biosynthesis through the hexosamine pathway even as overall glucose uptake declines. This may represent a compensatory stress response: metabolically stressed neurons loading their proteins with additional glycan modifications to stabilize them under conditions of energy deficit. The response may be protective in the short term but becomes pathological when sustained, creating a molecular environment increasingly inhospitable to the neuronal maintenance the brain depends on. This glucose-hexosamine-glycan axis may be a second downstream consequence of the same upstream metabolic failure that impairs amyloid clearance.
Reducing glycan biosynthesis through two independent approaches improved memory in AD mice. A genetic tool reducing the activity of PGM3, the enzyme producing the primary glycan building block, and a pharmacological inhibitor of the glycan assembly machinery both reduced brain glycan levels and improved social recognition memory performance. Crucially, neither produced significant changes in amyloid plaque burden or tau pathology over the two-week experimental period, suggesting the cognitive benefit operates through a mechanism independent of the protein pathology that existing drugs target.
Increasing glycan availability through oral glucosamine at the human therapeutic equivalent of approximately 2,500 milligrams per day worsened cognitive outcomes in AD mice but produced no measurable effect in healthy mice. Glucosamine-treated AD mice showed essentially no social recognition across all four trials of the memory test, a pattern more severely impaired than untreated AD mice. Healthy mice showed no changes in brain glycan levels or memory performance at the same dose, consistent with the healthy brain having homeostatic mechanisms that buffer against glucosamine-driven glycan elevation that the AD brain lacks.
In a retrospective analysis of over 50,000 patients, glucosamine use for at least one year was associated with a 25 percent higher mortality risk over ten years in patients with established Alzheimer's disease-related dementia, and a 25 percent higher rate of progression from mild cognitive impairment to Alzheimer's disease. Approximately 8 percent of patients in each diagnostic group had documented glucosamine use, a consistency across groups suggesting the identification method was reliable. No significant mortality association was observed in MCI patients, paralleling the mouse model finding that the harmful effect is specific to more severely affected brains. The human evidence is observational and cannot establish causality, but its directional consistency with the experimental findings makes it worth taking seriously.
The glucose-hexosamine-glycan axis connects this study to a broader landscape of metabolic interventions with potential neuroprotective relevance. SGLT2 inhibitors lower circulating blood glucose and raise ketone bodies, with BHB rising from approximately 0.14 to 0.27 mmol/L in non-diabetic individuals, potentially engaging the glycan pathway from two directions simultaneously. Dietary carbohydrate restriction, intermittent fasting, and exercise all reduce glucose flux through the hexosamine pathway through mechanisms directionally consistent with limiting glycan overproduction. None of these connections have been directly tested in the context of brain glycan metabolism, but they provide a more specific molecular rationale for the neuroprotective effects of metabolic interventions than prior research has been able to offer.
Introduction: Another Piece of the Alzheimer's Metabolic Puzzle
The history of Alzheimer's drug development is, in important ways, a history of a single hypothesis pursued with extraordinary commitment and extraordinary expense. The amyloid cascade hypothesis, which holds that the accumulation of amyloid-beta protein in the brain is the initiating event from which every other feature of Alzheimer's disease follows, has guided the field for three decades. It has generated hundreds of clinical trials, billions of dollars of investment, and a long and dispiriting record of failures. Drugs that successfully reduced amyloid burden in treated patients did not reliably produce cognitive benefit. The hypothesis survived each failure with modifications and refinements, but the clinical record has forced a reckoning that is now reshaping how the field thinks about what Alzheimer's disease actually is and where its causal origins lie.
The reckoning has been productive. What has emerged from the wreckage of the amyloid trials is not a vacuum but a growing recognition that Alzheimer's disease is a metabolic disease as much as a protein aggregation disease, that the amyloid accumulation the field has focused on may be as much a consequence of metabolic failure as a cause of neurodegeneration, and that the pathways most upstream of the clinical disease have not yet been adequately characterized or therapeutically targeted. The evidence for this metabolic reframe has been accumulating across multiple research programs. Fluorodeoxyglucose PET imaging has established that glucose uptake in the brain declines years before any clinical symptom of Alzheimer's disease appears, suggesting that the metabolic disruption precedes rather than follows the cognitive decline. Mitochondrial dysfunction creates the energy deficit that impairs the cellular machinery responsible for clearing amyloid-beta, providing a mechanistic link between metabolic failure and amyloid accumulation rather than the other way around. And a series of studies examining the lipid environment, inflammatory signaling, and vascular biology of the AD brain have each added dimensions to a picture of a brain progressively losing its metabolic homeostasis in ways that create conditions permissive to the protein pathology that ends in dementia.
A new paper published in Nature Metabolism from Ramon Sun's laboratory at the University of Florida adds a chapter to this story that the field has not been reading. It identifies hyperglycosylation, the excessive accumulation of complex sugar-protein modifications called glycans throughout the Alzheimer's brain, as a metabolic driver of the disease. The finding did not emerge from a targeted hypothesis about glycan biology in neurodegeneration. It emerged from an unbiased spatial analysis of the entire metabolic landscape of the AD brain, which is what made the glycan signal, when it appeared, genuinely surprising and genuinely significant. Using state-of-the-art imaging technology that can map the distribution of thousands of molecules simultaneously across intact brain tissue, the team identified dramatically elevated glycan levels across both grey and white matter in human Alzheimer's brains. They established that this hyperglycosylation is driven by increased glycan biosynthesis rather than impaired clearance. They demonstrated that reducing glycan biosynthesis genetically improves cognitive outcomes in AD mice, and that increasing it through oral glucosamine supplementation worsens them. And they showed, in a retrospective analysis of over 50,000 patients, that glucosamine use was associated with a 25 percent higher mortality risk in patients with established Alzheimer's disease-related dementia and a 25 percent higher rate of progression from mild cognitive impairment to Alzheimer's disease.
Glucosamine is one of the most widely used dietary supplements in the world, taken primarily by older adults for joint health. The population most likely to be using it is also the population most at risk for Alzheimer's disease. Understanding what this study found, how it was established, and what it does and does not mean for that population requires understanding the biology of glycosylation in the brain, why its disruption matters for neurological function, and how the specific metabolic pathway the study identifies connects to the broader landscape of Alzheimer's disease biology that prior research has been mapping.
What Glycosylation Is and Why It Matters for Brain Health
To understand why hyperglycosylation in the Alzheimer's brain is biologically significant, it helps to start from what glycosylation actually is and what it is doing in the brain under normal conditions. Glycosylation is not a peripheral or incidental process. It is one of the most fundamental and widespread modifications the cell makes to its proteins, and the brain depends on it more heavily than almost any other organ.
Proteins Do Not Work Alone
Every protein in the body is built from a sequence of amino acids encoded by its gene, but the protein that emerges from the ribosome is rarely the final functional product. After translation, proteins undergo a series of chemical modifications that alter their shape, stability, activity, and interactions with other molecules. These post-translational modifications are the cell's primary mechanism for creating functional diversity from a limited genetic code: the same protein can perform different roles in different contexts depending on what modifications it carries.
Glycosylation is among the most complex and information-rich of these modifications. It involves the attachment of complex carbohydrate chains, called glycans, to specific sites on a protein's surface. These glycan chains are not simple sugars but elaborate branching structures built from multiple sugar units assembled in precise sequences, creating molecular shapes that other proteins can recognize and interact with. A single protein may carry multiple glycan chains, each potentially different in structure, and the combination of glycan modifications on a protein constitutes a molecular identity that determines how it behaves in the cellular environment.
The attachment of N-linked glycans, the class of glycan modifications central to this study, occurs in the endoplasmic reticulum and Golgi apparatus, the intracellular assembly and processing centers where proteins are built, folded, modified, and routed to their destinations. As a protein moves through this processing system, glycans are added, trimmed, and elaborated in a precise sequence of enzymatic steps that produces the final glycan profile the protein will carry throughout its functional life. This process is not optional. Proteins that cannot be properly glycosylated are often misfolded, unable to reach their correct cellular locations, or targeted for degradation before they can function. Glycosylation is woven into the fundamental architecture of how proteins are made, quality-controlled, and deployed throughout the cell.
Why the Brain Depends on Glycosylation
The brain is one of the most glycan-rich organs in the body. This is not a coincidence. The extraordinary functional complexity of the brain, its dependence on precisely timed, spatially specific molecular interactions between hundreds of cell types communicating through thousands of different signals, places an exceptional demand on the molecular recognition systems that glycosylation provides.
At the synapse, the junction between neurons through which information is transmitted across the brain, glycosylation is essential for the formation and maintenance of the molecular machinery that makes synaptic transmission possible. Neurotransmitter receptors, the proteins that receive the chemical signals released by one neuron and convert them into electrical responses in the next, are heavily glycosylated, and their glycan modifications influence how efficiently they respond to neurotransmitter binding, how long they remain at the synapse, and how they interact with the scaffolding proteins that organize the synaptic architecture. Synaptic plasticity, the ability of synapses to strengthen or weaken their connections in response to activity, which is the cellular basis of learning and memory, depends on glycan-mediated changes in the composition and organization of the synaptic proteome. Disruptions in synaptic glycosylation compromise the molecular substrate of memory formation itself.
Beyond the synapse, glycosylation plays a critical role in neuroimmune signaling, the communication between neurons and the immune cells of the brain that determines how the neural environment responds to stress, damage, and the threat of infection. Microglia, the brain's resident immune cells, rely on glycan recognition systems to identify cellular debris that needs to be cleared, pathogens that need to be neutralized, and synapses that need to be pruned. The glycan coat that surrounds cells, called the glycocalyx, is a molecular identity tag that communicates whether a cell is healthy or damaged, whether it should be protected or eliminated. When glycan modifications are disrupted, the immune surveillance and phagocytic functions of microglia are impaired, contributing to the accumulation of toxic proteins and cellular debris that characterizes the Alzheimer's brain.
The blood-brain barrier, the specialized vascular structure that regulates what molecules can enter the brain from the circulation, is maintained in part through glycan-dependent interactions between the endothelial cells that line brain blood vessels and the astrocytes and pericytes that surround and support them. Glycocalyx dysregulation in brain endothelial cells has been shown to impair blood-brain barrier integrity in aging and disease, allowing inflammatory molecules from the periphery to enter the neural environment more freely and contributing to the neuroinflammatory cascade that drives neurodegeneration.
Glycosylation also regulates intracellular protein trafficking, the process by which proteins are routed to their correct destinations within the cell. In neurons, which have extraordinarily long axons and dendrites that require proteins to be transported over distances vastly greater than in most other cell types, the glycan modifications on a protein serve as molecular address labels that guide it to the synaptic terminal, the cell body, the dendritic spine, or whatever location its function requires. Misrouting of proteins through glycosylation errors can result in their accumulation in the wrong compartments, their failure to reach synapses that depend on them, or their interaction with proteins they would not normally encounter, all of which can compromise neuronal function and contribute to the toxic protein aggregation that defines Alzheimer's pathology.
What Happens When Glycosylation Goes Wrong
The importance of glycosylation for brain function is perhaps most clearly illustrated by what happens when the glycosylation machinery fails entirely. Congenital disorders of glycosylation are a group of genetic conditions caused by mutations in genes encoding glycosylation enzymes, and virtually without exception they produce severe neurological impairment. Patients with these disorders experience intellectual disability, epilepsy, cerebellar dysfunction, and abnormal brain development from birth, establishing that intact glycosylation is not optional for the brain to develop and function normally.
These congenital disorders represent the catastrophic failure of glycosylation. What the new study identifies is a more subtle but potentially equally consequential failure mode operating in the opposite direction: not the absence of glycosylation, but its excess. Hyperglycosylation, the accumulation of glycan modifications beyond what the brain's proteins normally carry, creates a different but potentially equally disruptive molecular environment. The proteins that carry excess glycan modifications may fold differently, traffic differently, interact with different molecular partners, and resist the clearance mechanisms that normally remove them when they become damaged or dysfunctional. Understanding why this excess accumulates in the Alzheimer's brain, and what it does when it does, is what the new study set out to determine.
The Hexosamine Biosynthetic Pathway: The Metabolic Engine of Glycosylation
Before examining what the study found, it is worth understanding the specific metabolic pathway that connects glucose metabolism to glycan biosynthesis, because this connection is central to understanding why the Alzheimer's brain, a brain characterized by glucose hypometabolism, might simultaneously be producing excess glycans.
The hexosamine biosynthetic pathway is a metabolic route that converts a fraction of the glucose entering the cell into the sugar building blocks needed for glycan assembly. Glucose enters the pathway and is converted through a series of enzymatic steps into N-acetylglucosamine, one of the primary sugar units used to build both N-linked glycans and other glycan modifications. The enzyme PGM3, one of the key targets in this study, catalyzes one of the critical conversion steps in this pathway, producing the activated form of N-acetylglucosamine that glycan biosynthetic enzymes in the endoplasmic reticulum and Golgi can use directly.
This pathway is normally regulated in proportion to the cell's overall metabolic state. When glucose is abundant, a modest fraction flows through the hexosamine pathway to support normal glycosylation. When glucose is scarce, the pathway's activity should, in principle, decline along with the overall glucose supply. What the new study suggests is that in the Alzheimer's brain, this proportional regulation is disrupted: even as overall glucose uptake declines, the fraction of available glucose being channeled through the hexosamine pathway toward glycan biosynthesis increases, creating a situation where glycan production is elevated even in a metabolically stressed brain. Understanding how and why this redirection occurs is one of the most important mechanistic questions the study opens.
What the new study suggests is that in the Alzheimer's brain, this proportional regulation is disrupted: even as overall glucose uptake declines, the fraction of available glucose being channeled through the hexosamine pathway toward glycan biosynthesis increases, creating a situation where glycan production is elevated even in a metabolically stressed brain.
The Discovery: The AD Brain Is Hyperglycosylated
The starting point for this study was not a targeted hypothesis about glycan biology in Alzheimer's disease. It was an unbiased look at the entire molecular landscape of the AD brain using a technology capable of mapping thousands of different molecules simultaneously across intact brain tissue while preserving their precise spatial locations. That methodological choice, to look broadly before looking narrowly, is what made the glycan finding possible and what makes it credible as a genuine discovery rather than a confirmation of a prior assumption.
The Technology: Reading the Brain's Molecular Geography
The imaging technology at the center of this study is called matrix-assisted laser desorption ionization mass spectrometry imaging, or MALDI-MSI. The name is technical but the concept is not too difficult to understand. A tissue section is placed on a surface coated with a chemical matrix, and a laser fires at different positions across the tissue in a precise grid pattern. At each position the laser causes molecules in the tissue to become ionized, acquiring an electrical charge that allows them to be detected and identified by a mass spectrometer based on their mass-to-charge ratio. By firing the laser at thousands of positions across the tissue section and recording which molecules are present at each position, the technology produces spatial maps of molecular distribution that can reveal where specific metabolites, lipids, or glycans are concentrated, depleted, or altered across different anatomical regions of the tissue.
The University of Florida team used an enhanced version of this approach that allowed them to analyze metabolites, lipids, and glycans from the same tissue section in a single experiment, a technical achievement that significantly reduces the analytical variability that comes from comparing separate experiments on separate tissue samples. They applied this multiplexed spatial multiomics approach to human frontal cortex samples from Alzheimer's disease patients and healthy controls matched for age, sex, and the time between death and tissue collection, a matching that is important because post-mortem biological changes can confound comparisons between patient and control samples.
What They Found in Human Brain Tissue
The metabolomics and lipidomics data revealed a range of molecular alterations in the AD samples, consistent with what prior research had established about the metabolic and lipid environment of the Alzheimer's brain. But the glycomics data produced a finding that stood out as unexpected in both its magnitude and its consistency. Glycan abundance was dramatically elevated across both grey and white matter regions in the AD brain samples. This was not a subtle or borderline elevation in one or two glycan species. It was a widespread and consistent increase across multiple glycan types, visible as a striking pattern of elevated signal throughout the spatial maps of the AD tissue compared to controls.
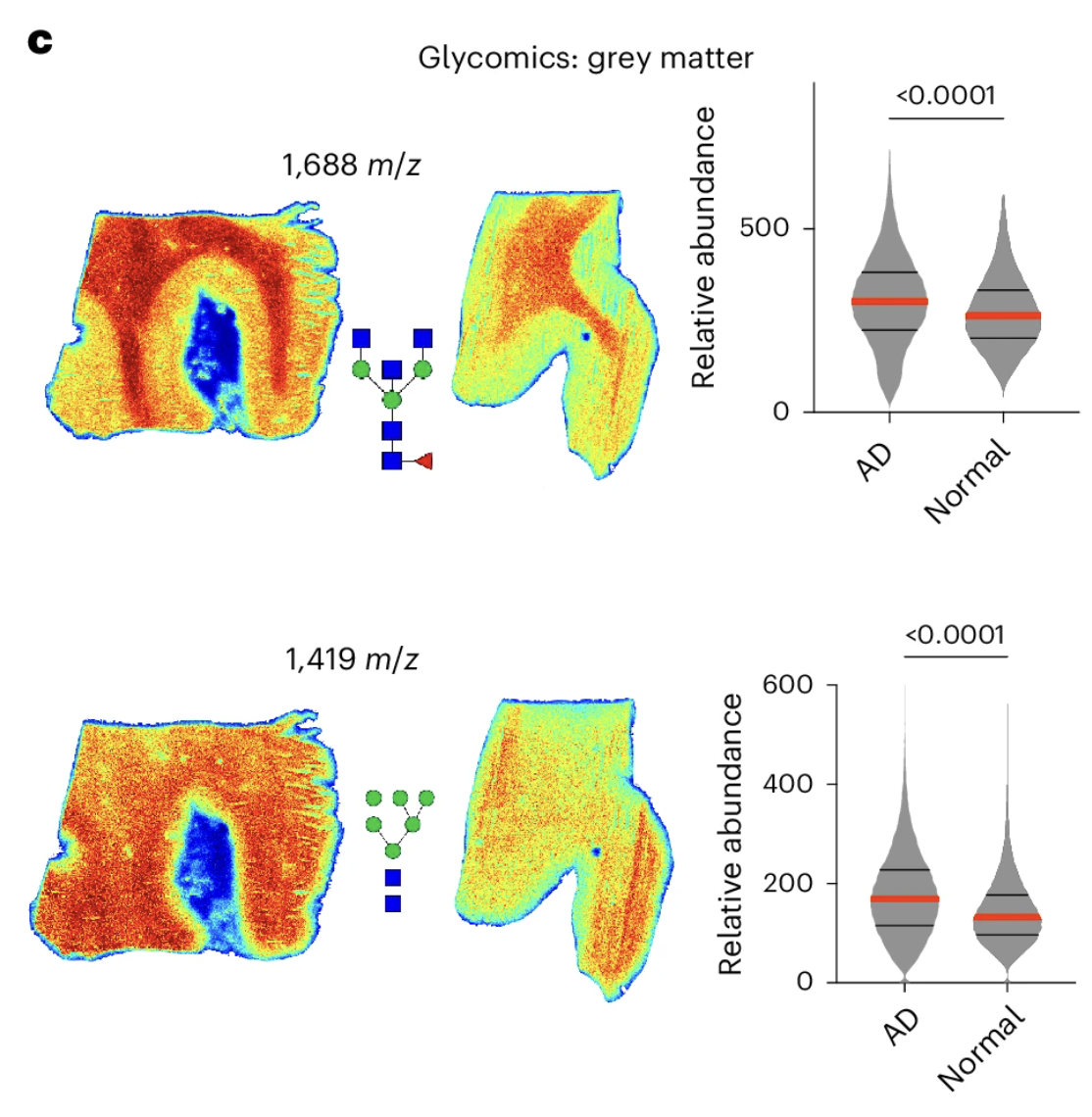
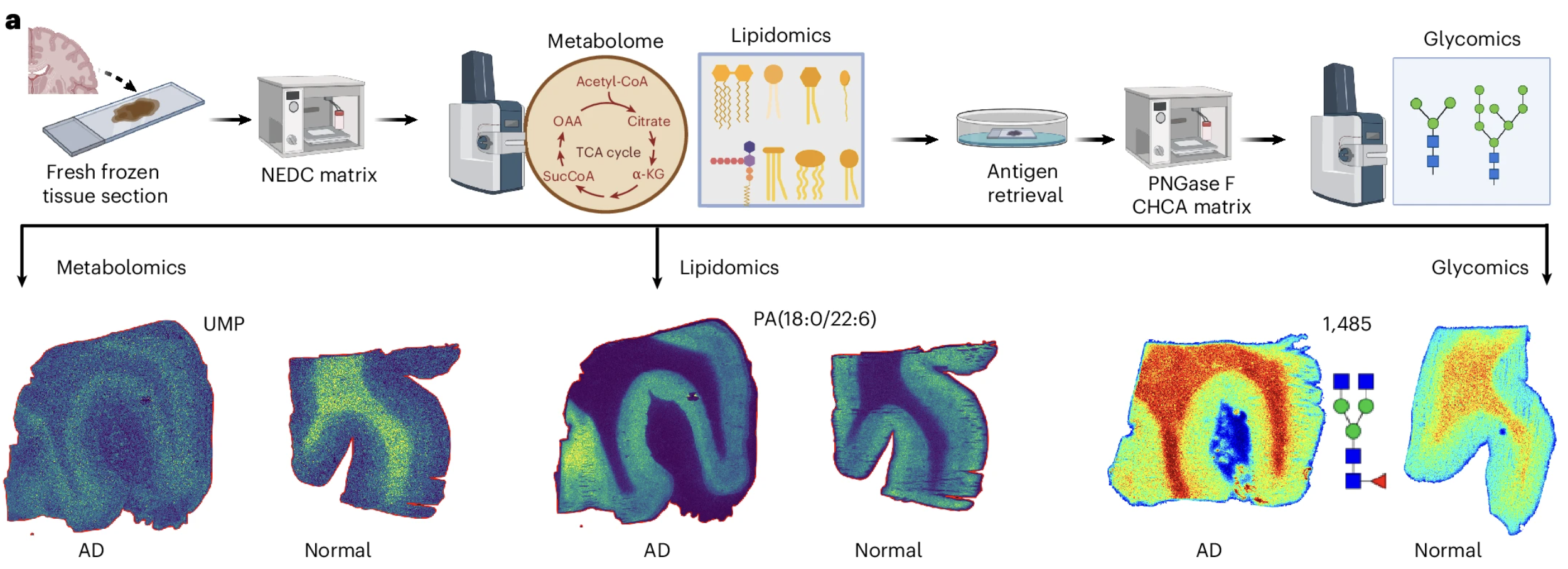
Figure 1: Spatial multiomics reveals widespread hyperglycosylation in human Alzheimer's brains. Panel A shows the imaging workflow. Panel C maps the spatial distribution of specific elevated glycan species across the tissue, illustrating how broadly the glycan overload is distributed.
The specific glycan species showing the most pronounced elevations included both bisecting glycans and high mannose glycans, two structurally distinct classes of N-linked glycans that serve different functional roles in the cellular glycoprotein landscape. High mannose glycans represent earlier, less processed forms in the glycan maturation pathway, while bisecting glycans represent more elaborately processed structures associated with specific immune and signaling functions. The elevation of both classes simultaneously suggests that the hyperglycosylation in the AD brain is not a specific defect in one step of glycan processing but a broader upregulation of the glycan biosynthetic program across multiple stages.
Alongside the elevated glycan levels, the spatial metabolomics data revealed a significant reduction in two critical glycan precursors: N-acetylglucosamine and glucosamine-6-phosphate, the building block molecules that the hexosamine biosynthetic pathway produces for glycan assembly. This finding is metabolically informative. The depletion of glycan precursors in the same tissue that shows elevated glycan levels is consistent with a biosynthetic process running faster than usual, consuming its substrate molecules at an elevated rate and leaving the precursor pool depleted as a consequence. It is not consistent with a simple accumulation of glycans due to impaired clearance, which would not be expected to deplete the biosynthetic precursors in this way.
Progressive Hyperglycosylation Across Disease Stages
The finding in a small matched cohort of three AD and three control samples, while striking, required validation in a larger and more carefully characterized patient series before its biological significance could be fully assessed. The team assembled a broader cohort of human AD brain samples stratified by Braak stage, the pathological staging system used in Alzheimer's disease that classifies disease severity based on the anatomical extent of tau pathology, from stage zero indicating no pathological changes through stages five and six indicating the most severe and widespread disease.
The Braak staging analysis produced a finding that strengthened the case for hyperglycosylation as a genuine feature of Alzheimer's disease biology rather than a coincidental observation in a small sample. In grey matter, glycan abundance increased progressively across Braak stages, with the most pronounced elevations in the later stages corresponding to the most severe disease. The relationship between glycan elevation and disease stage was not a threshold effect that appeared only in advanced disease but a gradual progression visible across the full range of stages, suggesting that hyperglycosylation tracks the advancing pathology of Alzheimer's disease as it spreads through the brain rather than appearing abruptly at a late stage.
White matter showed a different pattern: glycan levels elevated in early Braak stages but did not continue to increase through the later stages. This divergence between grey and white matter hyperglycosylation is biologically interesting and suggests that the cellular populations and molecular mechanisms driving glycan accumulation may differ between these two brain compartments, with grey matter neurons and their associated glial populations showing a sustained and progressive relationship with glycan elevation that white matter structures do not maintain through the full disease course.
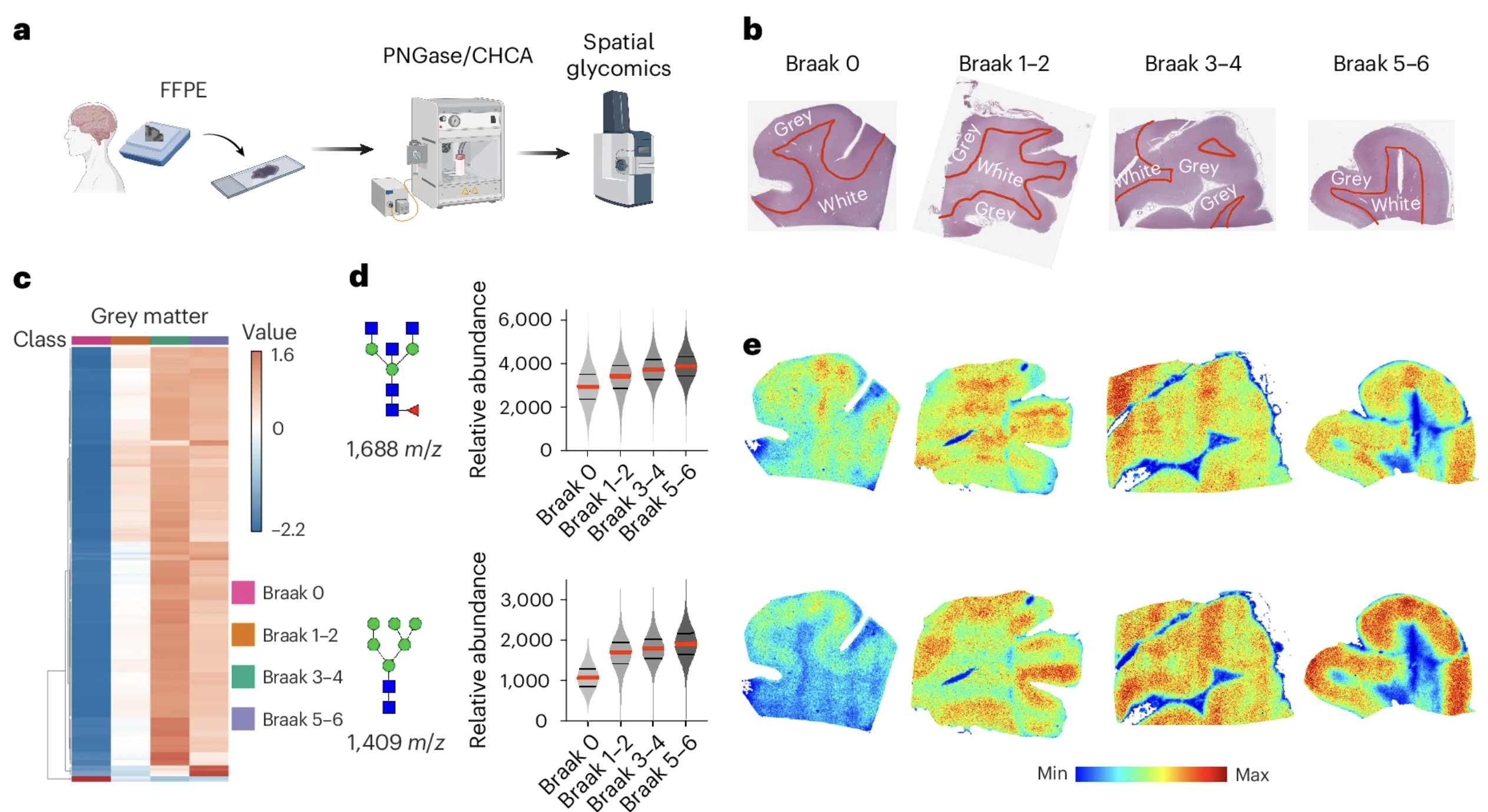
Figure 2: Glycan accumulation in grey matter increases progressively with Alzheimer's disease severity. The heatmap in Panel C and violin plots in Panel D show glycan abundance rising consistently across Braak stages from zero through five and six, tracking the advancing pathology of the disease. Panel E shows spatial glycan distribution maps across Braak stages, with glycan signal intensity visibly intensifying in the regions most affected by advancing Alzheimer's pathology.
Together, these human findings established something the field had not previously characterized: that the Alzheimer's brain carries a systematically elevated glycan burden across its grey matter that progresses with disease severity, visible in the spatial molecular landscape of the tissue in a way that post-translational proteomics and conventional biochemical assays had not captured. The next question was whether this phenomenon was specific to human Alzheimer's disease or reflected a conserved biological response to neurodegeneration that could be studied mechanistically in animal models.
Hyperglycosylation Is Conserved Across Species and Model Systems
A finding in human post-mortem brain tissue, however striking, carries inherent limitations as a basis for mechanistic investigation. Establishing that hyperglycosylation is a conserved feature of Alzheimer's pathology in well-characterized animal models was therefore an essential step toward understanding its biological origins and functional significance.
The team examined two widely used transgenic mouse models representing distinct pathological pathways. The 5xFAD model drives aggressive amyloid-beta accumulation, while the PS19 model drives tau pathology and neurofibrillary tangle formation. Together they cover the two major pathological hallmarks of Alzheimer's disease through distinct genetic mechanisms, making a shared phenotype across both models considerably more informative than the same observation in a single model.
At nine months of age, a stage corresponding to significant established disease in both models, the team applied the same spatial multiomics analysis used on human tissue. Both models showed the same widespread hyperglycosylation phenotype observed in human AD brains, with elevated N-glycan levels across multiple brain regions. The glycan elevation was most pronounced in the cortex, hippocampus, and thalamus, precisely the regions most critically involved in memory and cognitive processing that Alzheimer's disease systematically impairs. The cerebellum and hindbrain showed less pronounced hyperglycosylation, consistent with the regional vulnerability pattern of clinical disease progression.
The appearance of hyperglycosylation in both an amyloid model and a tau model establishes that the phenomenon is not a consequence of any single pathological protein but a common feature of the neurodegeneration that both cascades produce. Whether it is driving that neurodegeneration or reflecting it is the question the study's subsequent experiments address directly.
Glycoproteomic analysis of both human and mouse AD samples confirmed that neurons were the primary cell population undergoing enhanced glycosylation, connecting the hyperglycosylation phenomenon directly to the cells whose loss defines clinical disease severity. Importantly, more than 90 to 95 percent of the glycoproteins identified in AD samples were also present in normal samples. What has changed is not which proteins are glycosylated but how heavily they are glycosylated, a quantitative dysregulation of a normal biological process rather than an entirely aberrant one. The target is a specific excess rather than a foreign molecular event.
The proteins carrying excess glycans in the AD brain are the same neuronal membrane proteins that are normally glycosylated for synaptic transmission and action potential propagation. Their hyperglycosylation represents a quantitative dysregulation of a normal biological process rather than the emergence of an entirely aberrant one, which is both mechanistically informative and therapeutically relevant: the target is a specific excess rather than a completely foreign molecular event.
Why Hyperglycosylation Is Happening: Increased Biosynthesis Not Impaired Clearance
When any biological molecule accumulates in excess, the cause falls into one of two categories: too much is being produced, or too little is being cleared. These two mechanisms point toward entirely different therapeutic strategies, so establishing which one is driving hyperglycosylation in the AD brain matters enormously for understanding what to do about it.
The Isotope Tracing Experiment
To distinguish between these possibilities, the University of Florida team used stable isotope tracing, a technique that tracks the flow of material through metabolic pathways by replacing a common atom with a heavier isotopic form that creates a distinctive mass signature detectable by mass spectrometry. Mice were fed a diet enriched with a heavy carbon isotope version of glucose. As the mice metabolized this labeled glucose, the heavy carbon atoms were incorporated into glycans built from it, allowing the team to directly measure how fast new glycans were being synthesized and how fast existing ones were being cleared.
The experiment ran in two phases. In the pulse phase, mice consumed the labeled diet for 24 hours, allowing newly synthesized glycans to accumulate the heavy carbon label. In the chase phase, a separate cohort consumed the labeled diet and then switched back to an unlabeled diet for 24 hours, allowing the labeled glycans to be cleared while any newly synthesized glycans during the chase period would be unlabeled. The pulse phase measures production. The chase phase measures clearance.
The Result: Biosynthesis Is the Driver
The result was unambiguous. During the pulse phase, 5xFAD mice incorporated significantly more heavy carbon label into their glycans than wild-type controls, indicating elevated biosynthesis across multiple glycan species. During the chase phase, there was no significant difference in glycan turnover between the two groups. Clearance was normal. The hyperglycosylation in the Alzheimer's brain is being driven by making more glycans, not by failing to clear them. The therapeutic target is the production side of the equation.
What the experiment establishes is that the specific excess of glycans observed in the AD brain is attributable to increased production rather than impaired degradation, which is the mechanistically relevant distinction for understanding hyperglycosylation as an independent pathological driver.
Confirming the Biosynthetic Upregulation
Two additional lines of evidence reinforced this conclusion. The molecular building blocks that glycan production consumes were significantly elevated in AD mice, consistent with a biosynthetic process drawing down its raw materials at an elevated rate. And the genes encoding the enzymes responsible for assembling glycans showed elevated expression in both human AD brains and AD mouse models, confirming that the glycan production program had been broadly upregulated at the level of gene expression.
One nuance is worth noting. The sugar molecule that feeds into N-linked glycan production also feeds two other biological processes in the brain. If the entire supply of this molecule was being elevated uniformly, all three downstream processes should increase together. Instead, the team found that the other two processes were actually reduced in AD mice. The AD brain appears to be specifically redirecting its available sugar resources toward N-linked glycan production at the expense of competing pathways, a selective metabolic reprogramming rather than a simple across-the-board increase in biosynthetic activity.
The Metabolic Connection: Where the Extra Glycan Material Is Coming From
The finding that glycan biosynthesis is elevated in the Alzheimer's brain raises a question that sits at the intersection of glycan biology and the broader metabolic hypothesis of Alzheimer's disease. Where is the raw material for this elevated glycan production coming from, and why would a brain that is metabolically stressed and glucose-deprived be channeling its limited resources toward making more glycans?
The Glucose Paradox
One of the most reproducible findings in Alzheimer's disease research is that the AD brain takes up less glucose than a healthy brain, detectable on FDG-PET imaging years before any clinical symptom appears. The conventional interpretation is straightforward: the AD brain is energy-starved, and the resulting deficit impairs neuronal signaling, synaptic maintenance, and cellular quality control.
This is almost certainly correct as far as it goes. But the new study suggests it is incomplete. A brain taking up less glucose overall may nonetheless be channeling the glucose it does receive differently, with a larger fraction flowing toward glycan biosynthesis and a smaller fraction reaching the energy production pathways neurons depend on most. The depletion of glycan biosynthetic precursors, the elevated expression of glycan biosynthetic enzymes, and the selective redirection of hexosamine metabolism toward N-linked glycan production are all consistent with a brain that has shifted its metabolic priorities in a way that amplifies glycan production at the expense of energy generation.
A Compensatory Response Gone Wrong
Why would the AD brain make this shift? The authors offer a hypothesis that is biologically coherent. Under metabolic stress, cells sometimes attempt compensatory responses that are protective in the short term but become pathological when sustained. Increased glycosylation of neuronal proteins might represent such a response: an attempt by metabolically stressed neurons to stabilize their protein machinery and preserve cellular function under conditions of energy deficit by loading their proteins with additional glycan modifications.
Think of it as reinforcing a structure under stress by adding more bracing material. In the short term, the additional glycosylation might genuinely stabilize proteins becoming dysfunctional under metabolic pressure. But sustained hyperglycosylation changes the behavior of those proteins in ways that may ultimately compromise rather than preserve neuronal function, creating a molecular environment increasingly inhospitable to the cellular maintenance processes that neurons depend on for long-term survival.
This compensatory hypothesis makes sense of an otherwise puzzling observation: hyperglycosylation is occurring simultaneously with reduced glucose uptake rather than in spite of it. The metabolic stress of glucose hypometabolism may be triggering the upregulation of glycan biosynthesis as a stress response, one that once established may actively contribute to the neurodegeneration it was originally mobilized to counteract. In this framing, both pathways, impaired amyloid clearance and compensatory hyperglycosylation, emerge from the same upstream metabolic failure, which may partly explain why targeting amyloid alone has not been sufficient to produce consistent cognitive benefit.
What Reducing Glycosylation Does to AD Mice: The Genetic Evidence
The experiments described so far establish that the Alzheimer's brain is hyperglycosylated, that this hyperglycosylation is driven by elevated biosynthesis, and that it preferentially affects neurons through a selective redirection of the brain's metabolic resources toward glycan production. These are important mechanistic findings. But they do not answer the question that matters most for understanding whether hyperglycosylation is a therapeutic target: is the elevated glycan burden causing cognitive impairment, or is it simply accompanying a disease process driven by other mechanisms?
Answering a causal question requires manipulation. If hyperglycosylation is genuinely contributing to cognitive decline, then reducing it should produce cognitive improvement. If it is simply a bystander phenomenon that correlates with disease severity without driving it, then reducing it should leave cognitive function unchanged. The University of Florida team tested this prediction through two independent experimental approaches that targeted glycan biosynthesis through mechanistically distinct routes, providing a more robust causal test than either approach alone would allow.
The PGM3 Knockdown
The first approach targeted PGM3, the enzyme that produces the primary building block for glycan assembly. Reducing PGM3 activity constrains the rate at which glycan biosynthesis can proceed without disrupting the broader protein machinery that neurons depend on. Think of it as reducing the supply of raw material to a factory that has been overproducing: the factory keeps running, but at a lower output.
The team delivered a genetic tool directly into the brains of eight-month-old AD mice that reduced PGM3 production specifically, without affecting other proteins in the cell. Two weeks later, brain glycan levels were significantly reduced in treated mice. When those mice were tested on social memory, which measures whether a mouse recognizes a previously encountered individual by progressively reducing its interaction time across four trials, the treated AD mice showed meaningful improvement. Their recognition memory behavior more closely resembled that of cognitively healthy animals. Reducing glycan biosynthesis in the Alzheimer's brain was sufficient to improve a measure of memory function that the disease had impaired.
The NGI-1 Pharmacological Confirmation
The second approach used a drug called NGI-1 that targets a different step in the glycosylation process, blocking the molecular machinery that physically attaches glycan chains to proteins rather than reducing the supply of building blocks. A single injection produced reduced brain glycan levels and improved social memory performance in AD mice two weeks later.
The convergence of two mechanistically distinct interventions on the same cognitive improvement substantially strengthens the causal argument. Different tools, different molecular targets, same disease model, same result. The probability that this reflects coincidence rather than a genuine relationship between hyperglycosylation and cognitive impairment is low.
What Did Not Change: The Amyloid Finding
One of the most important observations in the intervention experiments is what the glycan-targeting treatments did not do. Despite the reduction in brain glycan levels and the improvement in social memory, neither the PGM3 knockdown nor the NGI-1 treatment produced significant changes in amyloid plaque burden or tau pathology over the two-week experimental period.
This matters for two reasons. First, the cognitive benefit cannot be explained by a reduction in the protein pathology the field has focused on. The brain was not less diseased by conventional measures. It was functioning better through a mechanism that appears independent of amyloid and tau, pointing toward glycan-dependent changes in synaptic function or neuroinflammatory signaling as the more likely explanation.
Second, this amyloid-independence is potentially significant for how glycan-targeting approaches might eventually be used clinically. Drugs that successfully clear amyloid have not reliably improved cognition in human trials. A treatment that improves cognition through a parallel and independent pathway could, if it translates to humans, be used alongside amyloid-targeting therapies rather than as a replacement for them.
Specificity to the Diseased Brain
When the same PGM3 knockdown was applied to healthy mice rather than AD mice, it produced no measurable change in social memory performance despite modestly reducing glycan levels. The cognitive benefit of reducing glycan biosynthesis appears to be specific to the disease context where glycan levels are already abnormally elevated. The therapeutic goal is not to eliminate glycosylation but to bring the pathologically elevated glycan burden of the AD brain back toward normal, and the experimental evidence suggests that this restoration produces cognitive benefit without harm in a brain that has not been pushed below the healthy baseline.
What Increasing Glycosylation Does to AD Mice: The Glucosamine Evidence
If reducing glycan biosynthesis improves cognitive outcomes in AD mice, the mirror prediction follows directly: increasing glycan biosynthesis should worsen them. The team tested this prediction using glucosamine, a supplement that most people associate not with Alzheimer's disease but with joint health, and the result was consistent with the prediction in a way that has immediate implications for how this widely used supplement should be thought about in the context of neurodegeneration.
Why Glucosamine
Glucosamine is a natural compound found in the body and is one of the primary building blocks of glycans. As a dietary supplement, it is taken by millions of older adults, typically at doses of around 1,500 to 2,500 milligrams per day, to support joint cartilage health. Its use is widespread precisely in the demographic most vulnerable to Alzheimer's disease. What makes glucosamine particularly useful for this experiment is that it crosses the blood-brain barrier and incorporates directly into brain glycans, making it an ideal tool for testing the effect of increased glycan availability in the brain without genetic manipulation or pharmaceutical intervention.
The team administered oral glucosamine to eight-month-old 5xFAD mice at a dose calculated to be equivalent to the standard human therapeutic dose, adjusted for the metabolic differences between mice and humans. After two weeks of daily treatment, spatial glycomics analysis confirmed that brain glycan levels were significantly elevated in glucosamine-treated AD mice compared to water-treated controls. The supplementation was doing what glucosamine's known biology predicts: increasing the availability of glycan building blocks in the brain and driving their incorporation into glycan structures on neuronal proteins.
What It Did to Cognition
The cognitive consequences were consistent with the causal prediction. Glucosamine-treated 5xFAD mice showed a further worsening of social memory performance compared to untreated AD mice. Where untreated AD mice already showed impaired social recognition, the glucosamine-treated mice showed essentially no recognition of previously encountered individuals across all four trials of the social memory test, a pattern representing more severe cognitive impairment than the disease alone had produced.
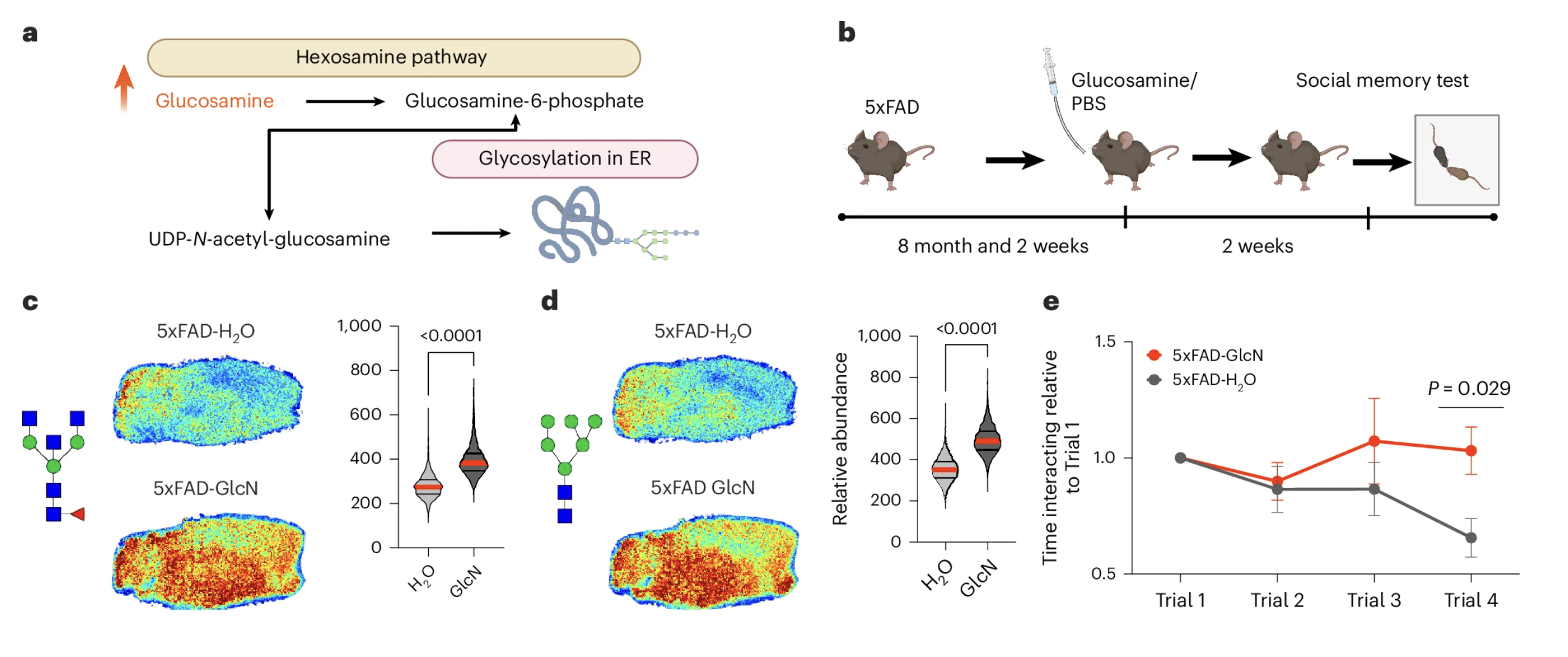
Figure 6: Glucosamine increases brain glycan levels and worsens social memory in Alzheimer's disease mice. Panels C and D show elevated brain glycan abundance in glucosamine-treated AD mice compared to vehicle-treated controls. Panel E shows social memory performance across four trials, with glucosamine-treated AD mice showing no recognition of previously encountered individuals across all trials, a pattern of more severe cognitive impairment than untreated AD mice displayed.
The parallel with the genetic experiments is clean and the argument is internally coherent. Reducing glycan biosynthesis through two independent genetic and pharmacological approaches improved cognition in AD mice. Increasing glycan availability through oral glucosamine worsened it. The cognitive consequences track the direction of the glycan manipulation in both directions, which is precisely what would be expected if hyperglycosylation is genuinely contributing to the cognitive impairment rather than simply accompanying it.
What Happened in Healthy Mice
The team administered the same glucosamine treatment to healthy wild-type mice at the same dose and duration. The result was striking in its contrast to the AD mouse finding. Glucosamine supplementation did not produce hyperglycosylation in the healthy mouse brain. Detailed glycomics analysis showed no significant changes in N-glycan abundance across multiple glycan species, and social memory performance was indistinguishable from vehicle-treated controls.
This is a critical observation for understanding the biological specificity of the glucosamine effect. The healthy brain appears to have homeostatic mechanisms that buffer against the glycan-elevating effect of exogenous glucosamine, maintaining normal glycosylation levels despite the increased substrate availability. The AD brain, which has already upregulated its glycan biosynthetic machinery and disrupted the regulatory balance that normally constrains glycan production, lacks this buffering capacity. When glucosamine provides additional raw material to a biosynthetic system that is already running above its normal set point, the result is a further elevation of an already elevated glycan burden. When it provides the same additional material to a system operating normally, homeostatic regulation absorbs the excess without producing measurable change.
This biological specificity of the glucosamine effect to the diseased brain is important context for the human evidence that follows. It suggests that the potential harms of glucosamine supplementation in the context of neurodegeneration are not a universal property of the supplement but reflect the specific metabolic vulnerability of the Alzheimer's brain to additional glycan substrate. The healthy brain, and by extension the cognitively healthy individual, appears to be better equipped to handle the glycan-elevating effects of glucosamine without consequence. Whether that resilience is maintained across the full spectrum from cognitive health through mild cognitive impairment to established dementia, and at what point the protective buffering capacity begins to fail, are questions that the mouse model data cannot definitively resolve and that the human evidence addresses in a different way.
The Human Evidence: Glucosamine in the Real World
The mouse model findings establish a causal relationship between glucosamine-driven hyperglycosylation and worsened cognitive outcomes in AD mice. But translating from mouse to human requires independent evidence. The University of Florida team sought that evidence in the electronic health records of the UF Health system, and what they found was consistent with the animal data in ways that are clinically significant even with the important limitations that observational human data carries.
The Dataset
The team identified over 50,000 patients diagnosed with Alzheimer's disease-related dementias and over 41,000 patients with mild cognitive impairment. To identify glucosamine users, they used physician notes and prescription records to find patients with documented glucosamine use of at least one year following diagnosis. Approximately 8 percent of patients in each diagnostic category had documented glucosamine use, a consistency across groups that suggests the identification method was capturing supplement use in a reproducible and unbiased way.
The Survival Finding
Ten-year survival analysis in the ADRD cohort showed that glucosamine use was associated with a 25 percent increase in mortality risk, with a p-value of 0.0023. In the MCI cohort, the same analysis showed no significant difference in mortality between glucosamine users and non-users. This divergence parallels the mouse model finding that glucosamine worsened outcomes in AD mice but not in healthy wild-type mice, suggesting that the harmful association may be specific to individuals whose neurodegeneration has progressed beyond a certain threshold rather than being a property of the supplement across all stages of cognitive aging.
The Disease Progression Finding
Among MCI patients with documented glucosamine use, the rate of transition to full dementia was 25 percent higher than among matched non-users. This connects the glucosamine signal to disease progression rather than simply to mortality, and suggests the biological effect may begin manifesting at the transition between mild impairment and established dementia, an earlier stage than the mortality finding alone would imply.
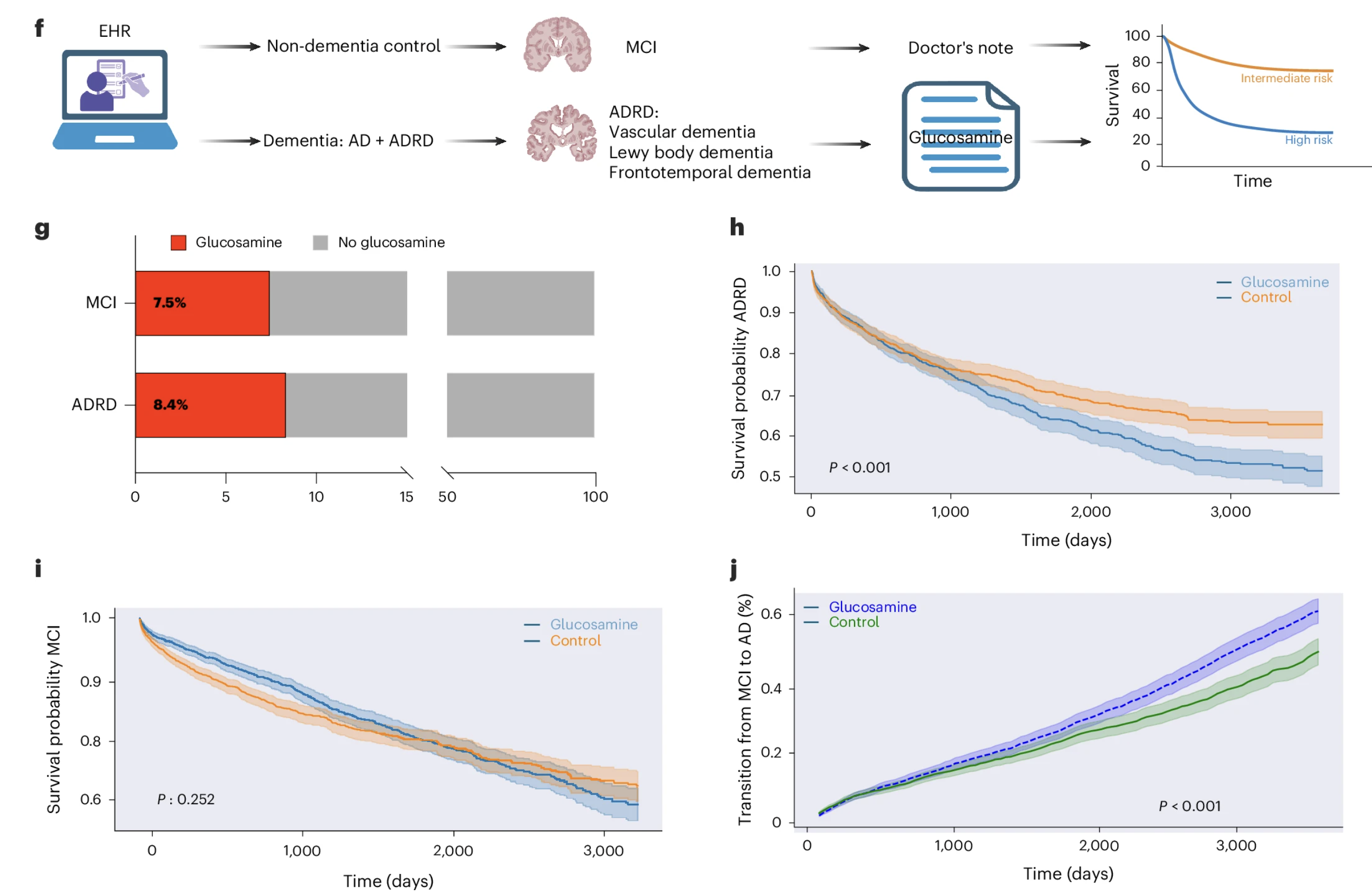
Figure 6: Glucosamine use is associated with worse outcomes in established dementia but not in mild cognitive impairment. ADRD patients using glucosamine showed reduced ten-year survival and MCI patients showed accelerated progression to dementia, while MCI survival was unaffected, consistent with the disease-specific vulnerability observed in the mouse model.
What the Human Evidence Can and Cannot Establish
The findings are observational and retrospective. They identify associations but cannot establish with certainty that glucosamine caused these outcomes. Glucosamine users may differ from non-users in ways that statistical adjustments did not fully capture, and physician note-based identification of supplement use has inherent limitations. What makes the evidence worth taking seriously despite these limitations is its directional consistency with the experimental findings across multiple analytical layers. The mouse models establish a causal mechanism. The isotope tracing establishes the biosynthetic driver. The genetic and pharmacological interventions establish the functional relationship. And the human data shows associations in the expected direction across two independent clinical outcomes in a dataset large enough to detect meaningful signals. No single element is definitive in isolation. Together, they form a coherent and internally consistent picture that warrants serious attention even before more controlled human studies have been conducted.
What This Does and Does Not Establish: Honest Limitations
A study that produces findings as practically significant as this one deserves careful scrutiny of its boundaries. The evidence assembled across mouse models, isotope tracing, genetic and pharmacological interventions, and electronic health records is coherent and internally consistent. It is also bounded in ways that matter for how confidently its conclusions should be held.
The Mouse Model Problem
The causal evidence for hyperglycosylation as a driver of cognitive impairment comes from mouse models representing familial Alzheimer's disease driven by specific mutations that account for a small fraction of human cases. Whether hyperglycosylation plays the same causal role in late-onset sporadic Alzheimer's disease, which accounts for the overwhelming majority of human cases, remains to be established. The two-week intervention window is also considerably shorter than the timescale over which human Alzheimer's disease develops. The absence of changes in amyloid plaque burden and tau pathology over this period establishes that the cognitive benefit is not mediated by rapid changes in plaques or tangles, which is mechanistically informative, but leaves open what longer-term glycan reduction might do to protein pathology. The social memory test measures one dimension of cognition relevant to Alzheimer's disease but does not capture the full spectrum of cognitive impairment the disease produces.
The Human Evidence Is Observational
The EHR analysis cannot establish causality. The 25 percent increase in mortality among ADRD patients using glucosamine and the 25 percent higher rate of MCI-to-ADRD progression are associations consistent with the causal story the mouse model data tells, but consistency is not proof. Two specific confounding factors deserve acknowledgment. Individuals with more severe or rapidly progressing Alzheimer's disease may be more likely to seek out supplements including glucosamine, creating reverse causation that cannot be fully excluded. And patients with more severe inflammatory arthritis, who would be more likely to use glucosamine consistently, may have elevated systemic inflammatory burden that independently worsens outcomes. The authors' requirement for at least one year of documented glucosamine use following diagnosis was designed in part to address the first concern, but it does not fully resolve either.
The limitations point directly toward the studies needed to advance the field. Prospective clinical studies examining glucosamine use and cognitive outcomes in well-characterized cohorts would address the confounding concerns of the retrospective EHR analysis. Studies examining hyperglycosylation in late-onset sporadic Alzheimer's disease would establish the translational relevance of the mouse findings to the form of the disease most people develop. Longer-term animal studies would clarify whether glycan-targeted approaches affect amyloid and tau pathology over extended timescales. Development of non-invasive biomarkers capable of measuring brain glycan burden in living humans would enable clinical monitoring in ways that post-mortem tissue analysis cannot. And brain-penetrant small molecule inhibitors of glycan biosynthetic enzymes, deliverable orally rather than through direct brain injection, represent the translational drug development challenge that would need to be solved before glycan-targeted interventions could reach human clinical trials.
Geroprotective and Lifestyle Interventions to Potentially Target Glycosylation
The identification of the hexosamine biosynthetic pathway as a driver of hyperglycosylation in the Alzheimer's brain opens an interpretive lens through which several established and emerging longevity interventions take on new mechanistic significance. None of what follows has been directly tested in the context of brain glycan metabolism and Alzheimer's disease, and the connections drawn here are mechanistically plausible but speculative. They are worth stating because they generate testable hypotheses and because they connect this study's findings to interventions that the broader longevity field is already engaged with.
The Glucose-Hexosamine Connection
The hexosamine biosynthetic pathway is glucose-sensitive. The rate at which it converts glucose into glycan building blocks scales with glucose availability, which means that anything reducing glucose flux through cellular metabolism has the potential to reduce the substrate supply for glycan biosynthesis. The connection between glucose overload and the hexosamine pathway is not a new observation. Healthspan's prior research review on glucose and ketones in Alzheimer's disease noted explicitly that oxidative stress drives glucose into alternative metabolic routes including the hexosamine pathway as a toxic detour that generates harmful intermediates in neurons. The hyperglycosylation study now provides the downstream consequence of that diversion: the hexosamine pathway is not simply producing toxic intermediates but is feeding an upregulated glycan biosynthetic program that actively contributes to cognitive impairment. The two findings are mechanistically complementary. Together they suggest that interventions targeting the glucose-hexosamine-glycan axis may be addressing a more specific and more consequential metabolic pathway in Alzheimer's disease than the general glucose hypometabolism framing alone would imply.
Dietary Interventions: Carbohydrate Restriction and Intermittent Fasting
Dietary approaches that reduce glucose availability, including low carbohydrate diets, ketogenic diets, and intermittent fasting protocols, reduce flux through the hexosamine pathway by limiting the glucose substrate that feeds it. Under conditions of carbohydrate restriction or fasting, the brain shifts toward alternative fuel sources including ketone bodies, reducing its dependence on glucose-derived energy and simultaneously reducing the glucose available for diversion into glycan biosynthesis. This does not mean that these dietary interventions have been shown to reduce brain hyperglycosylation in Alzheimer's disease. They have not. What it means is that the metabolic changes they produce are directionally consistent with reducing the substrate supply for the pathological process this study identifies, providing a plausible additional mechanism through which metabolic dietary interventions might support brain health alongside their established effects on insulin sensitivity, inflammation, and mitochondrial function.
Exercise and Metabolic Health
Regular exercise improves insulin sensitivity and glucose metabolism in ways that reduce chronic elevation of glucose flux through peripheral and central metabolic pathways including the hexosamine pathway. The metabolic improvements that exercise produces in the brain, including enhanced glucose uptake efficiency and improved mitochondrial oxidative capacity, could theoretically reduce the fraction of available glucose being diverted toward glycan biosynthesis by ensuring that more of it reaches the energy production pathways that compete with the hexosamine pathway for the same glucose supply. The relationship between exercise, brain glucose metabolism, and Alzheimer's disease has been studied extensively. The glycan pathway represents a potential additional dimension of this relationship that has not yet been investigated directly.
SGLT2 Inhibitors: A Dual Mechanistic Rationale
SGLT2 inhibitors are among the most compelling geroprotective drugs currently in clinical use, with established cardiovascular and renal benefits, emerging evidence for reduced all-cause mortality, and a growing body of observational evidence linking their use to reduced dementia risk. Their primary mechanism is the inhibition of glucose reabsorption in the kidney, which lowers circulating blood glucose and reduces the glucose load available to tissues throughout the body including the brain. They also produce a modest but sustained elevation of circulating ketone bodies, with BHB rising from approximately 0.14 to 0.27 mmol/L in non-diabetic individuals and from 0.24 to 0.56 mmol/L in diabetic individuals, a level of mild ketosis that prior research suggests may be sufficient to support mitochondrial function and reduce neuroinflammatory signaling in the brain.
Within the hyperglycosylation framework, SGLT2 inhibitors acquire a potential dual mechanistic rationale for neuroprotection that neither prior research nor this new study captures alone. The ketone elevation component may support neuronal energy metabolism and reduce neuroinflammation through the mitochondrial and anti-inflammatory pathways that prior research has established. The glucose reduction component may simultaneously reduce the substrate supply for the hexosamine pathway and glycan biosynthesis, addressing the upstream metabolic driver of hyperglycosylation through a mechanism that is already established as clinically safe and broadly beneficial. If both mechanisms are operating simultaneously, SGLT2 inhibitors would be engaging the glucose-hexosamine-glycan axis from two directions at once: reducing the glucose substrate available for glycan overproduction while simultaneously providing the brain with an alternative fuel source that does not feed the hexosamine pathway. This remains speculative and has not been directly tested in the context of brain glycan metabolism, but it is a mechanistically coherent hypothesis that the observational associations between SGLT2 inhibitor use and cognitive outcomes are consistent with and that purpose-designed studies could evaluate directly.
Rapamycin and mTOR Inhibition
Rapamycin occupies a relevant position in this discussion through a connection that is less direct but worth noting. mTOR signaling intersects with the hexosamine biosynthetic pathway through shared upstream nutrient-sensing machinery. Elevated mTOR activity, which characterizes metabolically stressed and aged tissue, promotes anabolic biosynthetic programs including, potentially, the upregulation of glycan production. Rapamycin's inhibition of mTOR recalibrates this anabolic drive, reducing the biosynthetic pressure that chronically elevated mTOR imposes on cellular metabolism. Whether this recalibration extends specifically to the hexosamine pathway and glycan biosynthesis in the aging brain has not been directly investigated, but the mechanistic connection through shared nutrient-sensing and biosynthetic regulatory pathways makes it a plausible hypothesis that intersects with the broader longevity biology of mTOR inhibition.
The Honest Framing
The connections drawn in this section are mechanistically informed speculation rather than established findings. The hyperglycosylation hypothesis is new, the direct evidence that any of these interventions affects brain glycan metabolism in humans does not yet exist, and the field will need purpose-designed studies examining glycan endpoints alongside metabolic and cognitive outcomes before these connections can be evaluated with the rigor they deserve. What the hyperglycosylation finding provides is a new mechanistic framework through which the neuroprotective effects of metabolic interventions can be understood and investigated, one grounded in a specific and newly identified pathological pathway rather than in the general observation that metabolic health and brain health are linked. The glucose-hexosamine-glycan axis now represents a concrete molecular target for that investigation.
Conclusion: A Metabolic Driver That Has Been Hiding in Plain Sight
Alzheimer's disease research has spent three decades following amyloid. The drugs built on that pursuit have produced a long record of clinical failures that the field is only now fully reckoning with, and the reckoning has been productive: it has forced a broader examination of the metabolic, vascular, and cellular biology that surrounds and precedes the protein aggregation the field has focused on. What is emerging from that examination is a picture of Alzheimer's disease as a disorder of metabolic homeostasis as much as a disorder of protein folding, with multiple upstream metabolic failures converging on a neural environment that becomes progressively inhospitable to neuronal survival regardless of whether the amyloid it accumulates is the primary cause or a downstream consequence of that inhospitality.
The University of Florida study adds a specific and previously uncharacterized metabolic pathway to that picture. Glycan biosynthesis, the cellular process of building and attaching complex sugar chains to proteins, is dramatically upregulated in the Alzheimer's brain. That upregulation is not a passive accumulation of unclearable material but an active increase in production driven by elevated biosynthetic machinery operating across multiple enzymatic steps simultaneously. It preferentially affects neurons, the cells whose loss defines the clinical severity of the disease. It tracks disease progression across Braak stages in human tissue. It is conserved across two independent mouse models representing the disease's two major pathological drivers. And when it is reduced through genetic or pharmacological intervention, cognitive function improves, while when it is increased through oral glucosamine supplementation, cognitive function worsens, in animal models and in the direction predicted by the causal hypothesis in human observational data.
The glucosamine finding is the most immediately clinically resonant result in the paper, and it is worth stating clearly what it does and does not establish. It does not establish that glucosamine causes Alzheimer's disease or that healthy individuals taking it for joint health are at meaningful risk. The mouse model data and the human EHR findings both suggest that the harmful effect is specific to brains whose neurodegeneration has already disrupted the homeostatic mechanisms that normally buffer against glucosamine-driven glycan elevation. The healthy brain appears resilient to this perturbation in ways the diseased brain is not. What the evidence does establish is a coherent and internally consistent signal, across mouse models, pharmacological interventions, and human observational data, that in individuals with established Alzheimer's disease-related dementia or progressing mild cognitive impairment, glucosamine supplementation is associated with worse outcomes in ways that the biology of hyperglycosylation can now explain mechanistically.
The broader therapeutic implication of this study is not primarily about glucosamine avoidance. It is about the identification of a molecular target, glycan biosynthesis through the hexosamine pathway, that the Alzheimer's field has not previously been pursuing and that the available evidence now suggests is worth pursuing seriously. The genetic and pharmacological tools that reduced glycan biosynthesis in the mouse experiments are not ready for human use in their current form, but the identification of PGM3 and the oligosaccharyltransferase complex as candidate targets provides a starting point for the drug development effort that would be needed to translate these findings into clinical intervention. The interventions that are already in clinical use, including SGLT2 inhibitors, dietary approaches that reduce glucose flux through the hexosamine pathway, and exercise, all have mechanistic connections to the glycan biosynthesis pathway that this study illuminates, and that connecting tissue gives existing longevity interventions a more specific molecular rationale for their potential neuroprotective effects than prior research has been able to provide.
The metabolic hypothesis of Alzheimer's disease is not a single unified theory but a growing family of observations about how metabolic dysfunction at multiple levels, energy deficit, mitochondrial failure, glucose hypometabolism, lipid dysregulation, and now glycan overproduction, creates the biological conditions in which neurodegeneration progresses. Each new mechanistic finding adds a pathway to that family and opens new questions about how the pathways interact, which ones are most upstream, and which represent the most tractable therapeutic targets. The hyperglycosylation finding is a meaningful addition to that family. It is grounded in rigorous experimental work across multiple model systems and supported by human data whose limitations are real but whose directional consistency with the experimental findings is not coincidental. The glucose-hexosamine-glycan axis is now part of the Alzheimer's story in a way it was not before this study, and understanding it is part of understanding what the disease actually is and how it might eventually be stopped.
Related studies