Restoring the Brain's Mitochondrial Quality Control System Produced Protective Effects Across the Full Alzheimer's Disease Cascade
Prefer to listen? Hit play for a conversational, audio‑style summary of this article’s key points.
Alzheimer's disease has long been understood primarily as a disease of amyloid accumulation, but the amyloid hypothesis has produced a long record of drug trial failures. A growing body of evidence suggests that mitochondrial dysfunction and defective mitophagy may precede amyloid accumulation rather than follow it. When mitochondria fail and cellular energy falls, the machinery responsible for clearing amyloid-beta collapses. Amyloid may accumulate not because production has increased but because clearance has failed, making mitochondrial health a more upstream therapeutic target than the plaques themselves.
Most preclinical Alzheimer's research has been conducted in mouse models built around rare familial mutations that cause only one to two percent of human cases. This study used a more realistic model that allows human amyloid-beta to accumulate gradually under normal biological control, producing age-dependent cognitive decline and mitochondrial dysfunction that more closely resembles the disease that affects the overwhelming majority of human patients. Positive findings in this model are more informative than results from standard overexpression models because they are engaging biology that actually resembles the human disease.
Urolithin A is produced when gut bacteria metabolize polyphenols found in pomegranates, walnuts, and certain berries, and not everyone produces it efficiently. Its primary mechanism in Alzheimer's biology is enhancing mitophagy, the cellular process that identifies and clears damaged mitochondria before their dysfunction compounds energy failure. It also shifts the mitochondrial network toward the healthier, more connected morphology associated with efficient energy production. In prior cell culture work, urolithin A consistently outperformed three other mitophagy enhancers tested against the same Alzheimer's pathology.
EGCG, the primary bioactive compound in green tea, works through mechanisms that are distinct from and complementary to urolithin A's. It directly targets amyloid-beta aggregation, redirecting peptides away from toxic forms and converting existing toxic fibrils into less harmful ones. It crosses the blood-brain barrier, suppresses the chronic inflammatory activation of microglia, and enhances the clearance of phosphorylated tau. Where urolithin A works from within the mitochondrial system outward, EGCG reduces the amyloid burden and inflammatory environment that actively undermine what urolithin A is trying to restore.
Both treatments produced meaningful improvements in behavior across every test administered, with the combination consistently outperforming urolithin A alone. Treated mice showed better motor coordination, greater locomotor activity and exploratory behavior, improved spatial learning and memory, and a trend toward better working memory. The breadth of behavioral improvement across four distinct measures, each depending on different neural circuits, suggests the intervention was engaging a fundamental upstream process rather than producing a targeted effect on a single pathway.
At the molecular level, urolithin A corrected the mitochondrial fragmentation that characterizes the Alzheimer's brain, and the combination amplified this correction dramatically. The proteins that drive mitochondrial fragmentation were suppressed while the proteins that promote healthy mitochondrial connectivity were elevated, with the combination producing corrections roughly two to four times larger than urolithin A alone across most markers. One fusion protein showed a particularly striking difference, increasing more than four times as much with the combination as with urolithin A alone, suggesting genuine synergy between the two compounds rather than simply additive effects.
Mitochondrial function improved directly and measurably across three independent indicators. Oxidative stress fell, oxidative damage to cellular membranes fell, and energy output rose in both treatment groups. The combination produced greater improvements than urolithin A alone across all three measures. Taken together, the mitochondria in treated mice were running cleaner and producing more usable energy, which is the functional prerequisite for the downstream improvements in synaptic health and amyloid clearance that followed.
Synaptic gene expression showed some of the most striking differences in the dataset, particularly for the combination. One key synaptic protein increased more than seven times as much with the combination as with urolithin A alone, and several others showed similarly amplified responses. The physical structure of synaptic connections, measured directly through dendritic spine density and length, was significantly improved in both treatment groups, with the combination producing broader restoration across brain regions than urolithin A could achieve independently. These are the microscopic structures through which memory is encoded and retrieved, and their partial restoration is the structural correlate of the behavioral improvements the study measured.
The amyloid findings close the loop on the metabolic hypothesis. Both forms of amyloid-beta that accumulate in Alzheimer's disease were reduced in treated mice, with the combination producing the larger reduction. This is precisely the result the metabolic hypothesis predicts: restore the energy-dependent cellular machinery that clears amyloid, and amyloid levels fall as a downstream consequence. The intervention targeted the upstream mitochondrial failure and produced downstream effects on amyloid that approaches targeting amyloid directly have consistently failed to produce in the other direction.
Introduction: Alzheimer's Disease and the Metabolic Hypothesis
For decades, Alzheimer's disease has been understood primarily as a disease of protein accumulation. Amyloid-beta, a peptide fragment produced during normal cellular metabolism, misfolds and aggregates into plaques that accumulate between neurons. Tau, a protein that normally stabilizes the structural scaffolding inside neurons, becomes abnormally phosphorylated and forms tangles within them. The dominant hypothesis in the field, the amyloid cascade hypothesis, has held that this accumulation is the initiating event, the upstream cause from which everything else, the neuroinflammation, the synaptic damage, the cognitive decline, follows downstream. Drug development has followed this logic for more than two decades, producing a long and expensive series of trials targeting amyloid clearance that have largely failed to produce meaningful clinical benefit even when they successfully reduced amyloid burden.
The failures have forced a reckoning. If amyloid clearance does not reliably produce cognitive improvement, either the hypothesis is wrong, the interventions are arriving too late, or amyloid is not the upstream cause the field assumed it was. A growing body of evidence points toward the third possibility. Mitochondrial dysfunction, the failure of the cellular power plants that neurons depend on for their extraordinary energy demands, may not be a downstream consequence of amyloid accumulation. It may be an upstream driver of it. The brain's ability to clear amyloid-beta depends on energy. When mitochondria fail and cellular energy falls, the machinery responsible for processing and clearing amyloid-beta fails with it. Amyloid accumulates not because production has increased but because clearance has collapsed. In this framing, the plaques that have defined Alzheimer's disease pathologically for over a century are less the cause of the disease than a marker of a brain that has lost its metabolic footing.
Central to this metabolic vulnerability is a process called mitophagy, the cellular system responsible for identifying and clearing damaged mitochondria before they can compound the energy failure they represent. In a healthy cell, mitophagy functions like a quality control system for the power grid, continuously removing failing units before their dysfunction spreads. In the Alzheimer's brain, this system breaks down. Damaged mitochondria accumulate, generating oxidative stress, impairing energy production, and creating the conditions under which amyloid clearance collapses. Defective mitophagy is not merely a symptom of advancing Alzheimer's disease. It may be a core biological vulnerability that allows the disease to progress in the first place.
The researchers behind the paper published in Cells asked a consequential question within that framework. If defective mitophagy is a core vulnerability rather than a downstream consequence, can it be restored? And if so, can restoring it interrupt the cascade of cellular damage that ultimately produces the cognitive decline that defines the disease? To answer that question they turned to urolithin A, a compound produced when gut bacteria metabolize polyphenols found in pomegranates, and a combination of urolithin A with EGCG, the primary bioactive compound in green tea, testing both in a mouse model of late-onset Alzheimer's disease that more accurately reflects the form of the disease that affects the overwhelming majority of human patients.
What they found across behavior, mitochondrial biology, synaptic health, neuroinflammation, and amyloid levels is consistent with the metabolic hypothesis in ways that deserve careful attention, while remaining appropriately honest about the distance between a mouse model and a human disease.
The Mouse Model Problem: Why Most Alzheimer's Research Has Been Testing the Wrong Disease
To appreciate why the model used in this study matters, it helps to understand how most Alzheimer's mouse models were built and what assumptions are embedded in their design.
The genetic story of Alzheimer's disease has two very different chapters. The first involves rare inherited mutations in three genes, APP, PS1, and PS2, that cause a familial form of the disease with near-certain penetrance, striking affected individuals in their forties and fifties with a severity and speed that reflects the catastrophic impact of a single genetic lesion on the brain's ability to process amyloid-beta. This form of Alzheimer's is devastating and scientifically important, but it accounts for one to two percent of total cases. The people who develop it carry a specific mutation that can be identified, characterized, and modeled in animals with relative precision.
The second chapter involves everyone else. Late-onset sporadic Alzheimer's disease, which accounts for more than 98 percent of cases, does not arise from a single catastrophic mutation. It emerges from the interaction of genetic risk factors, most notably the APOE4 allele, with the accumulated cellular damage of biological aging, metabolic dysfunction, vascular changes, inflammatory history, and decades of environmental and lifestyle exposures that no genetic model can fully capture. It is a disease of aging biology rather than a disease of a specific gene, and it develops over timescales that compress awkwardly into any animal study.
The mouse models that have dominated preclinical Alzheimer's research were built around the first chapter. They overexpress mutant human APP, PS1, or PS2 genes, or combinations of these, in ways that produce amyloid pathology rapidly and dramatically. These models have been extraordinarily productive for understanding the molecular biology of amyloid accumulation and its downstream consequences. But they have also produced a long and dispiriting record of treatment failures: compounds that cleared amyloid beautifully in transgenic mice, restored cognitive performance on behavioral tests, and then did nothing meaningful in human clinical trials. The disconnect has become one of the central methodological problems in the Alzheimer's field, and it maps precisely onto the metabolic hypothesis. Models built around genetic overexpression of amyloid-producing mutations are designed to test the amyloid cascade hypothesis. They are not designed to test what happens when the brain's metabolic machinery begins to fail gradually across decades of biological aging. Testing a mitophagy-targeted intervention in a model built around amyloid overexpression is testing the wrong question in the wrong system.
The humanized amyloid-beta knockin model used in this study takes a fundamentally different approach. Rather than overexpressing a mutant human gene at artificially elevated levels, it replaces three specific amino acids in the mouse amyloid-beta sequence with their human equivalents, allowing the mouse to produce human amyloid-beta under normal regulatory control, expressed from the endogenous mouse APP gene at physiological levels. The result is a model where human amyloid-beta accumulates gradually and age-dependently, without the artificial acceleration that comes from overexpression, and where the cognitive decline, synaptic deterioration, and inflammatory changes that follow more closely resemble the slow progressive trajectory of late-onset sporadic Alzheimer's disease in humans.
These mice develop measurable behavioral impairments, mitochondrial dysfunction, synaptic damage, and rising amyloid-beta levels in an age-dependent fashion that begins showing clear pathological signs by seven months of age. Soluble amyloid-beta is detectable as early as three months and increases progressively, mirroring the gradual accumulation that precedes clinical symptoms in human late-onset AD by years or decades. They do not develop the explosive amyloid pathology of transgenic overexpression models. They develop something more modest and more honest, a biological deterioration that takes time to emerge, that involves the same mitochondrial dysfunction and metabolic failure that the metabolic hypothesis places at the center of the disease, and that more accurately represents the Alzheimer's disease that clinicians are actually trying to treat.
This matters for interpreting what urolithin A and the combination treatment produced in this study. The model is not asking whether a mitophagy enhancer can overcome the consequences of a catastrophic genetic overexpression of amyloid. It is asking whether restoring mitochondrial quality control in a brain undergoing the kind of gradual metabolic deterioration that drives late-onset Alzheimer's disease can interrupt the cascade that leads to cognitive decline. That is a more honest question, and the answer it produces is proportionally more informative.
What Goes Wrong in the Alzheimer's Brain: Mitochondria, Mitophagy, and the Cascade That Follows
The biological processes that deteriorate in Alzheimer's disease are interconnected in ways that matter for understanding why a mitophagy-targeted intervention might produce effects across so many different outcome measures simultaneously. Each process feeds the others, creating a cascade in which mitochondrial failure amplifies synaptic damage, synaptic damage compounds cognitive decline, and the inflammatory response to cellular dysfunction accelerates all of it. Understanding the cascade requires understanding each of its components, starting where the metabolic hypothesis places the initiating event.
The Brain's Extraordinary Energy Demand
The brain is the most metabolically demanding organ in the body. Despite representing roughly two percent of total body mass, it consumes approximately 20 percent of the body's total energy output at rest, almost entirely in the form of ATP generated by mitochondria. Neurons are particularly dependent on this continuous energy supply because unlike most cells in the body they cannot store meaningful energy reserves and cannot easily shift to alternative fuel sources when their primary supply is disrupted. The mitochondria distributed throughout a neuron's cell body, axons, and dendrites function as a distributed power grid, delivering ATP precisely where and when it is needed to maintain the electrical gradients that allow neurons to fire, the molecular machinery that allows synapses to transmit signals, and the quality control systems that keep the cellular environment clean and functional.
When this power grid begins to fail, the consequences are not limited to energy shortage. Mitochondria that are not functioning efficiently generate excessive reactive oxygen species, chemically aggressive molecules that damage proteins, lipids, and DNA in the surrounding cellular environment. They lose the electrochemical gradient across their inner membrane that drives ATP synthesis. They shift from the elongated, well-connected network morphology of healthy mitochondria toward a fragmented, disconnected state that is less efficient, more prone to further dysfunction, and harder to clear through the cell's quality control systems. Energy failure, oxidative stress, and structural deterioration compound each other in a cycle that, once established, is difficult to interrupt without addressing the underlying mitochondrial health problem directly.
Mitochondrial Dynamics: The Balance Between Fission and Fusion
Healthy mitochondria are not static structures sitting passively in the cell. They exist in a state of continuous dynamic remodeling, constantly dividing through a process called fission and merging through a process called fusion, with the balance between these two processes determining the overall health and efficiency of the mitochondrial network.
Fusion allows mitochondria to share components, dilute localized damage, and maintain the interconnected network morphology associated with efficient energy production. Fission allows the cell to segregate damaged portions of the network into discrete units that can be targeted for clearance before their dysfunction spreads. In a healthy cell, these processes are tightly regulated and balanced, with key proteins orchestrating each direction: Drp1 and Fis1 drive fission, while Mfn1, Mfn2, and Opa1 drive fusion.
In Alzheimer's disease, this balance shifts dramatically and consistently toward fission. Drp1 and Fis1 are elevated. Mfn1, Mfn2, and Opa1 are reduced. The mitochondrial network fragments into an excess of small, disconnected units that are less efficient at producing ATP, more prone to generating reactive oxygen species, and less capable of supporting the energy demands of active synapses. This fragmentation is not merely a morphological observation. It is a functional deterioration that compounds energy failure and creates a larger pool of damaged mitochondria in need of clearance through a mitophagy system that is itself becoming impaired.
Mitophagy: The Quality Control System That Fails
Mitophagy is the cellular process by which damaged or dysfunctional mitochondria are selectively identified, engulfed, and delivered to lysosomes for degradation and recycling. Think of it as the cell's dedicated waste disposal system for its power plants, a continuous quality control operation that prevents the accumulation of failing units from compromising the network as a whole.
The process begins when a mitochondrion loses sufficient membrane potential to trigger the activation of PINK1, a kinase that accumulates on the outer mitochondrial membrane of damaged units and recruits Parkin, an enzyme that tags the mitochondrion with molecular signals identifying it for clearance. A membrane structure called a phagophore then forms around the tagged mitochondrion, expands to enclose it completely in a double-membrane vesicle called an autophagosome, and fuses with a lysosome where the enclosed contents are enzymatically degraded. The resulting molecular components are recycled back into the cellular pool for use in building new, healthy mitochondria.
In Alzheimer's disease, this system is profoundly impaired. PINK1 and Parkin levels are substantially reduced in affected brain regions. The autophagosomal machinery that executes the clearance process is compromised. And amyloid-beta and phosphorylated tau interact directly with key mitophagy proteins in ways that further disrupt the process, creating a situation where the very toxic proteins whose accumulation defines Alzheimer's pathology are also actively undermining the cellular system responsible for maintaining the mitochondrial health that would otherwise limit their accumulation. This is one of the most compelling mechanistic arguments for placing mitochondrial dysfunction upstream of amyloid pathology rather than downstream of it: defective mitophagy creates the conditions for amyloid accumulation, and amyloid accumulation worsens defective mitophagy, establishing a self-reinforcing cycle that drives progressive neurodegeneration.
Mitochondrial Biogenesis: Replenishing the Network
Clearing damaged mitochondria through mitophagy is only half of the quality control equation. The cell also needs to continuously produce new, healthy mitochondria to replace those that have been cleared, a process called mitochondrial biogenesis regulated primarily by a protein called PGC-1 alpha, often described as the master regulator of mitochondrial production. PGC-1 alpha coordinates a transcriptional program that drives the expression of nuclear respiratory factors NRF1 and NRF2, which in turn activate TFAM, the mitochondrial transcription factor responsible for replicating the mitochondrial genome and initiating the production of new mitochondrial proteins.
In Alzheimer's disease, this biogenesis program is impaired. PGC-1 alpha, NRF1, NRF2, and TFAM are all reduced in affected brain regions, meaning the cell is not only failing to clear damaged mitochondria efficiently but also failing to replenish its mitochondrial network with healthy replacements. The result is a progressive depletion of functional mitochondrial capacity that compounds the energy failure driving the disease forward.
Synaptic Health: Where Mitochondrial Failure Becomes Cognitive Decline
The connection between mitochondrial dysfunction and cognitive decline runs through the synapse. Synapses are the connections between neurons through which information is transmitted across the brain, and they are among the most energy-intensive structures in the body. Maintaining the electrochemical gradients, molecular machinery, and structural components that allow synapses to transmit signals efficiently requires a continuous and reliable ATP supply. When mitochondrial function deteriorates and energy delivery to synapses becomes unreliable, synaptic transmission degrades.
The structural manifestation of this degradation is visible in dendritic spines, the microscopic protrusions on neuronal dendrites that receive incoming synaptic signals. Healthy neurons in brain regions associated with learning and memory maintain dense arrays of dendritic spines whose number, size, and morphology reflect the strength and activity of their synaptic connections. In Alzheimer's disease, dendritic spine density and length decline progressively, reducing the physical infrastructure of memory and learning in ways that correlate directly with cognitive impairment. Key synaptic proteins including synaptophysin and PSD95, which are essential for maintaining the structural integrity and functional efficiency of synaptic connections, are consistently reduced in affected brain regions.
Neuroinflammation: The Amplifier
Superimposed on the mitochondrial and synaptic deterioration is a chronic neuroinflammatory response that amplifies the damage rather than resolving it. Microglia, the brain's resident immune cells, respond to signals of cellular stress and damage including oxidative products of dysfunctional mitochondria, amyloid-beta aggregates, and dying neurons by shifting from a quiet surveillance state into an activated inflammatory state. In the context of acute injury this activation is protective, clearing debris and coordinating repair. In the context of chronic, diffuse cellular deterioration it becomes a sustained inflammatory response that damages healthy neurons alongside the dysfunctional ones, releases cytokines that impair synaptic function, and accelerates the neurodegeneration it was mobilized to address. Astrocytes, the supporting cells of the brain that regulate the synaptic environment and provide metabolic support to neurons, undergo a similar reactive transformation that further disrupts the neural environment.
Together these processes, mitochondrial fragmentation, energy failure, defective mitophagy, impaired biogenesis, synaptic deterioration, and chronic neuroinflammation, form the interconnected cascade that the metabolic hypothesis places at the center of Alzheimer's disease. They are not independent problems requiring separate solutions. They are a single system in progressive failure, and a compound that meaningfully engages the mitochondrial quality control process at the top of that cascade has the potential to produce effects that propagate across all of it. That is what urolithin A was designed to test, and what the combination with EGCG was designed to extend.
Two Compounds Worth Understanding: Urolithin A and EGCG
The two compounds at the center of this study were not chosen arbitrarily. Each has a specific mechanistic profile that maps onto a distinct point in the cascade described above, and the rationale for combining them is grounded in the complementarity of those profiles rather than in a general assumption that more intervention is better. Understanding what each compound does, and why their mechanisms are additive rather than redundant, is essential for interpreting what the combination finding means.
Urolithin A: A Mitophagy Enhancer From an Unexpected Source
Urolithin A is not consumed directly. It is produced. When the human gut microbiome metabolizes ellagitannins, a class of polyphenols found abundantly in pomegranates, walnuts, and certain berries, the resulting metabolites are converted by intestinal bacteria into a family of compounds called urolithins, of which urolithin A is the most biologically active. This dependence on gut bacterial conversion is one of urolithin A's most practically significant features: not everyone produces it efficiently. The capacity to convert dietary ellagitannins into urolithin A depends on the specific composition of the gut microbiome, which varies substantially between individuals, meaning that two people eating identical quantities of pomegranate can have dramatically different circulating urolithin A levels. This variability is one of the reasons supplemental urolithin A has attracted research interest as a more reliable delivery mechanism than dietary sources alone.
Urolithin A's primary mechanism of action in the context of Alzheimer's biology is the activation and enhancement of mitophagy. It acts specifically on the PINK1-Parkin pathway, the molecular system that identifies damaged mitochondria and initiates the process of tagging them for clearance. By enhancing the activity of this pathway, urolithin A effectively amplifies the cell's existing quality control signal, making it more sensitive to mitochondrial damage and more efficient at initiating the clearance process in response. It also promotes mitochondrial fusion over fission, shifting the dynamic balance of the mitochondrial network back toward the elongated, interconnected morphology associated with efficient energy production and away from the fragmented state that characterizes the Alzheimer's brain.
In earlier cell culture work by the same research group, urolithin A was tested against three other mitophagy enhancers, nicotinamide riboside, tomatidine, and actinonin, in hippocampal neurons transfected with mutant APP and mutant tau constructs. Across mitochondrial dynamics, biogenesis, mitophagy activation, synaptic gene expression, cell survival, and mitochondrial respiration, urolithin A consistently produced the strongest protective effects of any compound tested. It emerged from that comparison as the most potent mitophagy enhancer available for this biological context, which is what motivated its use as the primary intervention in the current mouse study and as the foundation of the combination treatment.
EGCG: Targeting Amyloid and Neuroinflammation From a Different Angle
Epigallocatechin gallate, EGCG, is the primary bioactive catechin in green tea and one of the most extensively studied polyphenols in the context of neurodegenerative disease. Its mechanisms are distinct from urolithin A's in ways that make the combination biologically coherent rather than redundant.
EGCG's most directly relevant property for Alzheimer's biology is its effect on amyloid-beta aggregation. It does not simply inhibit the formation of amyloid fibrils. It actively redirects amyloid-beta peptides away from the toxic fibrillar aggregation pathway toward alternative, less toxic molecular assemblies, and converts existing mature fibrils into smaller, non-toxic forms. This is a meaningfully different mechanism from the antibody-based amyloid clearance strategies that have dominated Alzheimer's drug development: rather than tagging amyloid for immune-mediated removal, EGCG alters the physical chemistry of amyloid assembly itself, preventing the formation of the structures that are most damaging to neurons and synapses.
EGCG crosses the blood-brain barrier, reaching the neural environment where Alzheimer's pathology develops rather than acting only in the periphery. Once in the brain it suppresses the inflammatory activation of microglia in response to amyloid-beta, reducing the production of pro-inflammatory cytokines including TNF-alpha, IL-1-beta, and IL-6 that amplify neuronal damage in activated microglial states. It modulates the oxidative stress environment in neural tissue through effects on endogenous antioxidant systems. And it has been shown to enhance the clearance of phosphorylated tau, the second major toxic protein in Alzheimer's pathology, adding a dimension of mechanistic breadth that extends beyond the amyloid biology.
Why the Combination Is More Than the Sum of Its Parts
The mechanistic logic for why urolithin A and EGCG might produce additive effects when combined is straightforward once both compounds' mechanisms are understood. Urolithin A is working from the inside of the cascade outward, restoring the mitochondrial quality control machinery that energy failure has compromised, shifting the dynamic balance of the mitochondrial network toward health, and creating the cellular conditions under which amyloid clearance can resume more effectively. EGCG is working from the outside of the cascade inward, reducing the amyloid burden that is driving mitochondrial dysfunction, suppressing the neuroinflammatory amplification that worsens it, and targeting the tau pathology that compounds both.
Together they are addressing the same disease from two directions simultaneously. Urolithin A is rebuilding the cellular maintenance system that Alzheimer's pathology has broken. EGCG is reducing the load that is doing the breaking. The combination does not simply double the mitophagy signal or double the amyloid suppression. It creates a more comprehensive intervention across the interconnected pathways that drive neurodegeneration, engaging the upstream metabolic vulnerability that the metabolic hypothesis places at the center of the disease while simultaneously reducing the downstream toxic burden that upstream failure produces. Whether that complementarity translates into synergistic rather than merely additive effects, and whether the specific doses and ratio used in this study are optimal, are questions the current data cannot fully answer. But the mechanistic rationale for the combination is coherent in a way that distinguishes it from combining two compounds simply because both have shown some benefit in isolation.
The Study: Design and What Was Measured
The experimental design of this study was built around a straightforward but demanding question: if defective mitophagy is a core vulnerability in late-onset Alzheimer's disease, does restoring it change the trajectory of the disease across the full range of outcomes that matter, from the molecular to the behavioral? Answering that question required measuring not just one or two endpoints but the entire cascade, from the proteins governing mitochondrial dynamics and quality control through the structural integrity of synapses and dendrites to the cognitive performance of living animals. The comprehensiveness of the measurement approach is one of the study's genuine strengths, and understanding what was measured and how is essential for interpreting what the findings mean.
The Animals and the Intervention
Three-month-old humanized amyloid-beta knockin mice were confirmed by genotyping and divided into three experimental groups. The first group served as untreated controls, receiving no intervention and providing the baseline picture of how the disease progresses in this model without interference. The second group received urolithin A alone, administered by intraperitoneal injection at a dose of 2.5 milligrams per kilogram of body weight three times per week. The third group received the combination of urolithin A at the same dose plus EGCG at 25 milligrams per kilogram of body weight, also by intraperitoneal injection three times per week. Both treatment protocols continued for four months, bringing the animals to seven months of age at the time of assessment, the point at which prior characterization of this model had established that behavioral impairments, mitochondrial dysfunction, synaptic damage, and rising amyloid-beta levels are reliably present and measurable.
The doses used for both compounds were established through prior optimization work in cell culture systems rather than chosen arbitrarily, which adds confidence that the concentrations being tested are in a biologically relevant range rather than pharmacologically extreme. Twenty animals per group were used for behavioral assessments, providing adequate statistical power for detecting differences in cognitive and motor performance. For the molecular, cellular, and structural analyses, five animals per group were used for each measurement approach, a sample size consistent with the field's norms for these technically demanding assays while acknowledging the statistical limitations that relatively small group sizes introduce.
The Behavioral Battery
Four behavioral tests were used to assess the functional consequences of treatment across different cognitive and motor domains, each designed to detect a specific dimension of the impairment that Alzheimer's pathology produces.
The rotarod test placed animals on a rotating cylinder that accelerated gradually over time, measuring how long each animal could maintain its balance before falling. This test assesses motor coordination and balance, functions that depend on cerebellar and motor cortical integrity and that deteriorate in hAbKI mice relative to wild-type controls. The open field test placed animals in a square arena for five minutes under moderate lighting and used video-tracking software to measure total distance traveled and average speed, providing an index of general locomotor activity and willingness to explore that is sensitive to both motor and anxiety-related changes. The Y-maze placed animals in a three-armed maze and measured the spontaneous alternation between arms, a behavior that reflects working memory and depends on intact hippocampal function. Animals with intact working memory tend to explore the arm they have not recently visited, while those with hippocampal impairment alternate less consistently. The Morris water maze placed animals in a circular pool of opacified water containing a hidden submerged platform and measured how efficiently animals learned to locate the platform across four days of training trials, providing a well-validated measure of spatial learning and long-term memory that is among the most hippocampus-dependent cognitive assessments available.
The Molecular and Cellular Assessment
After behavioral testing, brain tissues were collected and subjected to a comprehensive molecular and cellular analysis spanning multiple levels of biological organization. Quantitative real-time PCR measured mRNA expression levels for genes governing mitochondrial fission, fusion, biogenesis, mitophagy, autophagy, and synaptic function in cortical tissue, providing a readout of how gene expression programs had shifted in response to treatment. Immunoblotting analysis of cortical tissue protein extracts confirmed whether the gene expression changes translated into corresponding changes in protein levels for the same biological processes. Immunofluorescence imaging of hippocampal brain sections provided a spatially resolved visualization of how the protein changes distributed across the brain region most critical for memory and learning, confirming that the molecular changes observed in bulk tissue extracts were present in the hippocampal structures where Alzheimer's pathology most directly affects cognitive function.
Transmission electron microscopy of hippocampal and cortical sections provided direct visualization of mitochondrial morphology at a resolution that allows individual mitochondria to be counted, measured, and characterized as fragmented or elongated, and that allows mitophagosomal formations, the double-membrane structures through which damaged mitochondria are engulfed during the clearance process, to be identified and quantified. This level of structural analysis is technically demanding and not always included in preclinical Alzheimer's studies, and its presence here provides a direct visual confirmation of whether the mitophagy enhancement that urolithin A was expected to produce was actually occurring at the organelle level in the treated brain tissue.
Golgi-Cox staining of hippocampal and cortical neurons visualized dendritic spine density and morphology, providing a direct structural readout of synaptic health at the level of the physical connections between neurons. Sandwich ELISA measured soluble amyloid-beta 40 and amyloid-beta 42 levels in brain tissue homogenates, the two primary forms of amyloid-beta whose accumulation defines Alzheimer's pathology in this model. And mitochondrial functional assays measured three direct indicators of mitochondrial health in cortical tissue: hydrogen peroxide production as a marker of oxidative stress, lipid peroxidation as a marker of oxidative damage to cellular membranes, and ATP production as a direct measure of mitochondrial energy output.
Together these measurements span the full cascade from gene expression through protein levels through organelle morphology through cellular structure through tissue-level pathology through whole-animal behavior, creating a picture of how urolithin A and the combination treatment affected the Alzheimer's brain at every level accessible to the study's methods. It is that comprehensiveness, more than any single finding, that makes the results worth examining carefully.
What the Data Showed: From Molecules to Memory
The findings from this study are best understood not as a collection of independent results across different measurement domains but as a coherent story that unfolds across levels of biological organization, from the genes governing mitochondrial quality control through the physical structure of synaptic connections to the cognitive performance of living animals. That coherence is itself one of the most important features of the data: when an intervention produces consistent directional effects across molecular, cellular, structural, and behavioral outcomes simultaneously, it strengthens the case that the mechanism being targeted is genuinely relevant to the biology being measured rather than producing isolated effects in specific assays that do not connect to functional outcomes.
In every domain examined, urolithin A produced meaningful protective effects compared to untreated hAbKI mice. In every domain examined, the combination of urolithin A and EGCG produced stronger effects than urolithin A alone. The consistency of that pattern across seven distinct measurement domains is the central finding of the study, and it deserves to be read as a coherent signal rather than a collection of individual positive results.
Behavior: The Functional Readout
The behavioral findings are where the molecular and cellular story connects to something that would be recognizable in a clinical setting: an animal that moves better, explores more freely, learns more efficiently, and remembers more accurately. These are not abstract molecular endpoints. They are the functional consequences of what the disease does to the brain, and their partial reversal by treatment is the most clinically meaningful result in the dataset.
On the rotarod test, which measures motor coordination and balance by asking animals to stay on an accelerating rotating cylinder, urolithin A treated mice outperformed untreated controls significantly, reaching p equals 0.014. The combination treatment produced a stronger result at p equals 0.008. The numbers matter less than what they represent: treated animals were maintaining their balance longer, indicating that the neurological deterioration affecting motor circuits in untreated hAbKI mice was being partially reversed.
In the open field, which measures spontaneous locomotion and exploratory willingness, urolithin A treated mice covered more distance, p equals 0.022, and moved at higher average speed, p equals 0.037, than untreated controls. Reduced locomotion in hAbKI mice is not simply a motor problem. It reflects a broader reduction in motivated, exploratory behavior driven by the neurological changes Alzheimer's pathology produces. The combination again produced greater improvements across both measures.
The cognitive findings were particularly striking. The Morris water maze, which requires animals to learn and remember the location of a hidden platform using spatial cues, is one of the most hippocampus-dependent behavioral tests available, directly probing the memory circuitry most vulnerable to Alzheimer's pathology. Urolithin A treated mice found the platform significantly faster, p equals 0.013, and traveled significantly less distance in their search, p equals 0.020, compared to untreated hAbKI mice. The combination produced comparable improvements. In the Y-maze, both treatment groups showed trends toward improved working memory, measured by the tendency to explore novel maze arms spontaneously, though these did not reach statistical significance in either group. The direction of effect was consistent with the other behavioral findings even where the statistical threshold was not crossed.
The breadth of the behavioral improvements deserves emphasis. Motor coordination, locomotion, spatial memory, and working memory depend on distinct neural circuits and reflect different aspects of neurological health. An intervention that improves all of them simultaneously is not producing a targeted pharmacological effect on a single pathway. It is producing broad neurological benefit consistent with engaging a fundamental upstream process, which is precisely what the mitochondrial hypothesis predicts.
Mitochondrial Dynamics: Rebalancing Fission and Fusion
The molecular findings begin with the most direct test of whether urolithin A was doing what it was expected to do: correcting the balance between mitochondrial fission and fusion that Alzheimer's pathology shifts so profoundly toward fragmentation.
The mRNA fold change data captures this correction with quantitative precision. Recall that in untreated hAbKI mice, the fission machinery is overactive and the fusion machinery is suppressed, producing the excess of small, disconnected mitochondria that characterizes the Alzheimer's brain. In urolithin A treated mice, the fission gene Drp1 was suppressed by 1.96-fold and Fis1 by 2.32-fold, while the fusion genes moved in the opposite direction, Mfn1 increasing by 2.03-fold, Mfn2 by 3.4-fold, and Opa1 by 2.91-fold. The molecular tug-of-war between fragmentation and connectivity was shifting back toward health.
The combination produced larger corrections across every single marker. The Mfn1 increase is particularly striking: where urolithin A alone produced a 2.03-fold increase, the combination produced an 8.9-fold increase in the same gene, a fourfold amplification of the fusion signal from adding EGCG to the protocol. Mfn2 reached 5.23-fold with the combination compared to 3.4-fold with urolithin A alone, and Opa1 reached 3.76-fold compared to 2.91-fold. The synergy between the two compounds in restoring the fusion machinery is one of the most quantitatively dramatic findings in the dataset.
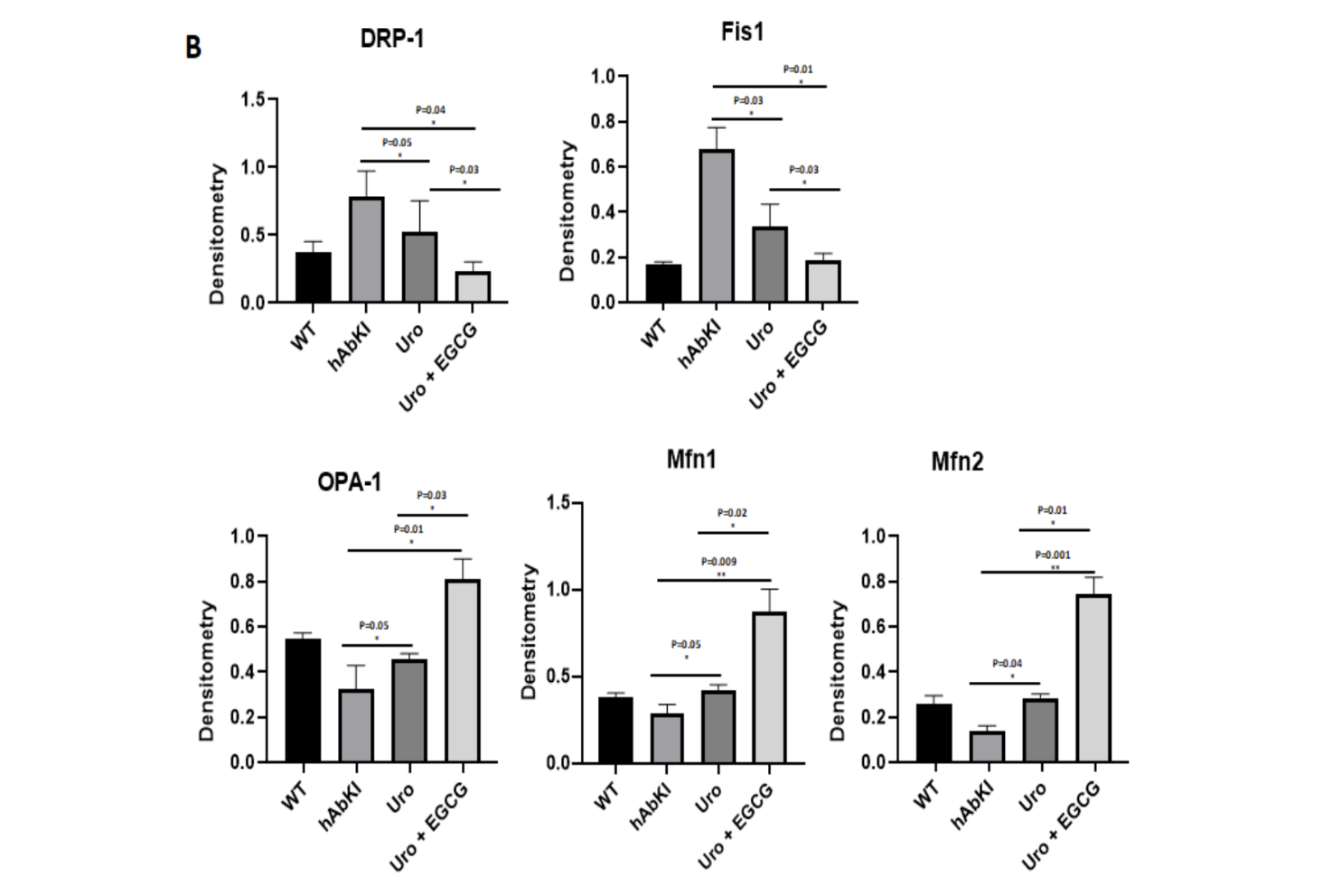
Figure 3. Mitochondrial fission proteins Drp1 and Fis1 were significantly reduced and fusion proteins Mfn1, Mfn2, and Opa1 were significantly increased in both treatment groups relative to untreated hAbKI mice, with the combination of urolithin A and EGCG producing consistently larger shifts than urolithin A alone across every protein measured.
Mitochondrial Biogenesis: Rebuilding the Network
Clearing damaged mitochondria is only half of the quality control equation. The cell also needs to replace what it has cleared with new, healthy mitochondria. This is the job of mitochondrial biogenesis, and restoring it alongside mitophagy is what distinguishes a comprehensive metabolic intervention from one that simply accelerates the removal of damaged units without rebuilding the network.
The biogenesis data shows that urolithin A was engaging both halves of the equation. PGC-1 alpha, the master regulator of mitochondrial production and one of the key proteins suppressed in Alzheimer's disease, increased by 3.03-fold in urolithin A treated mice. Its downstream targets Nrf1, Nrf2, and TFAM, which translate the PGC-1 alpha signal into the actual production of new mitochondrial proteins and DNA, increased by 1.46-fold, 2.39-fold, and 1.8-fold respectively. The biogenesis program was being activated at multiple levels simultaneously, not just in one regulatory protein.
The combination amplified these effects meaningfully. PGC-1 alpha reached 4.1-fold, Nrf1 2.76-fold, Nrf2 3.25-fold, and TFAM 2.2-fold. The picture at this level is of a mitochondrial network that is not simply being cleaned up but actively rebuilt, with the combination producing a more ambitious reconstruction program than urolithin A could accomplish alone.
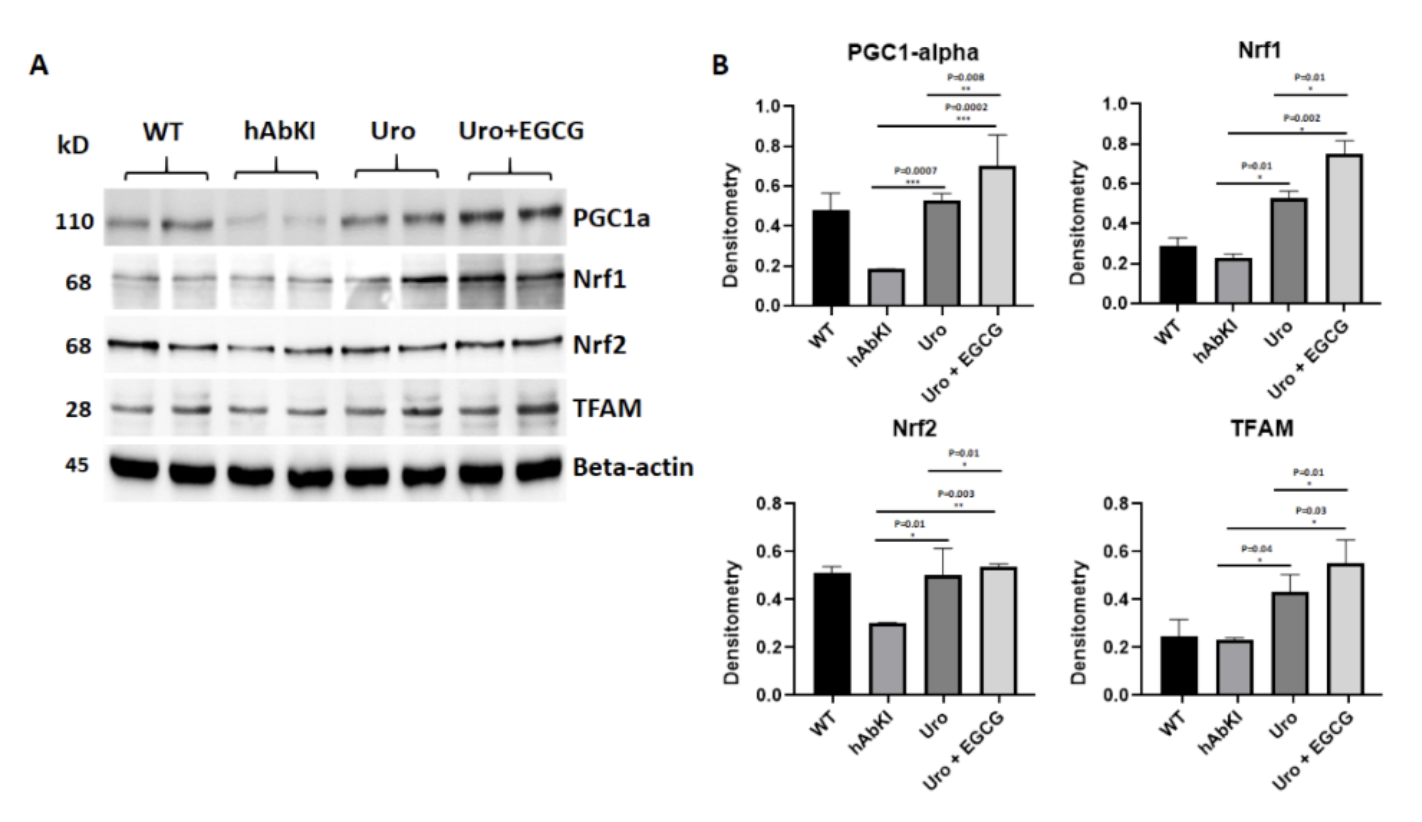
Figure 4: The mitochondrial biogenesis program suppressed in Alzheimer's disease is restored by both treatments. PGC-1 alpha, NRF1, NRF2, and TFAM were all significantly elevated, with the combination producing consistently larger increases across the entire biogenesis cascade.
Mitophagy and Autophagy: The Quality Control System Restored
The mitophagy findings provided the most direct confirmation that urolithin A was engaging its primary mechanism in the tissue that matters most for Alzheimer's disease. PINK1, which detects damaged mitochondria and initiates the tagging process, increased by 2.63-fold in urolithin A treated mice. Parkin, which executes the tagging, increased by 2.3-fold. These are the two proteins most consistently reduced in Alzheimer's disease brains, and their restoration signals that the quality control system is coming back online.
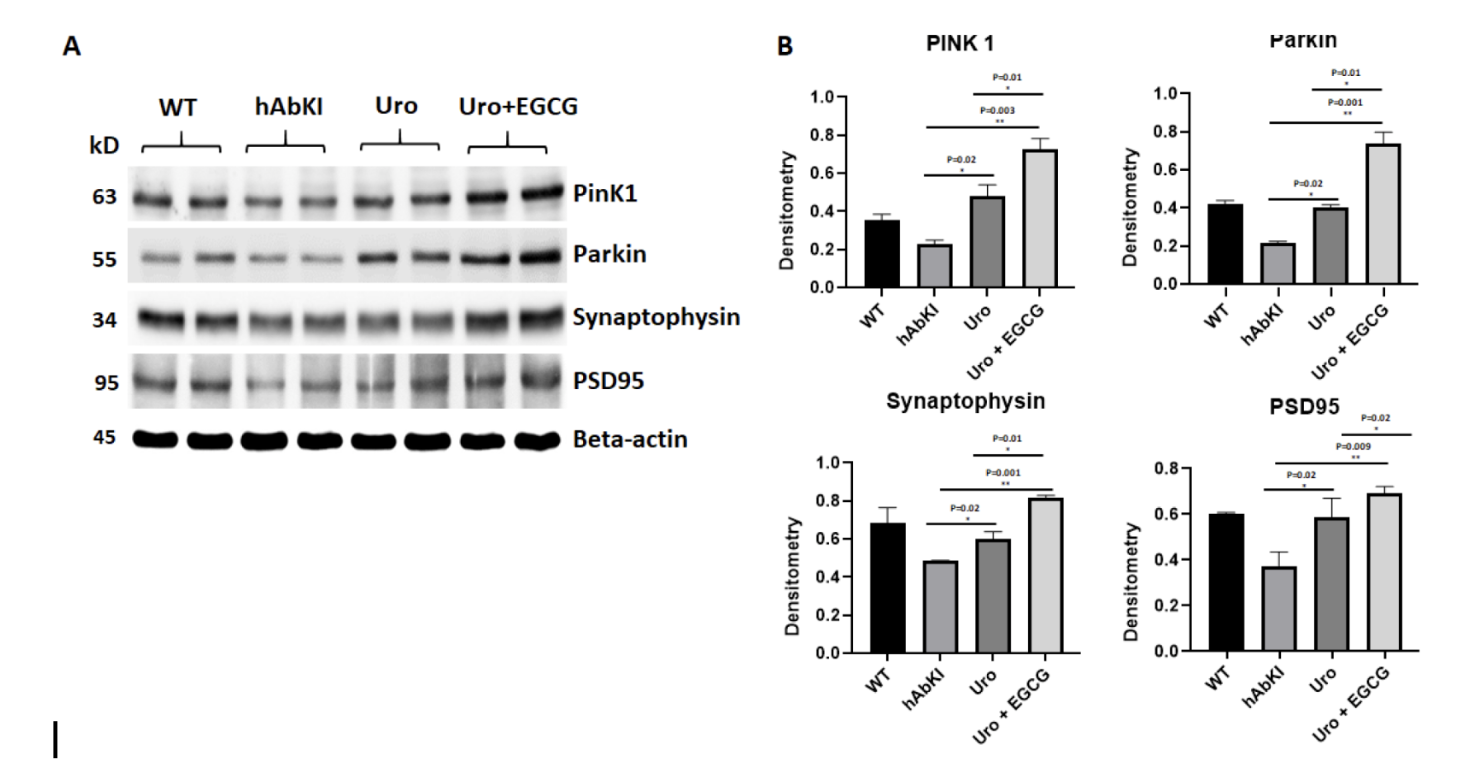
Figure 5: PINK1 and Parkin, the two proteins that initiate mitochondrial clearance and are consistently reduced in Alzheimer's disease, are significantly restored by both treatments.
The broader autophagy machinery, the molecular apparatus that physically engulfs tagged mitochondria and delivers them to lysosomes for degradation, showed equally significant improvements. Beclin, ATG5, LC3B-1, LC3B-2, and BCL2 were all significantly elevated in urolithin A treated mice, each reaching statistical significance between p equals 0.05 and p equals 0.0001. When the combination group was compared directly to urolithin A alone, the combination produced significantly higher levels of Beclin, ATG5, and BCL2, confirming that EGCG was amplifying the autophagy response beyond what mitophagy enhancement alone could produce. Transmission electron microscopy gave this molecular finding a physical face, showing more mitophagosomal formations in hippocampal tissue of treated mice, the actual double-membrane structures through which damaged mitochondria are engulfed, with more visible in the combination group than in the urolithin A group.
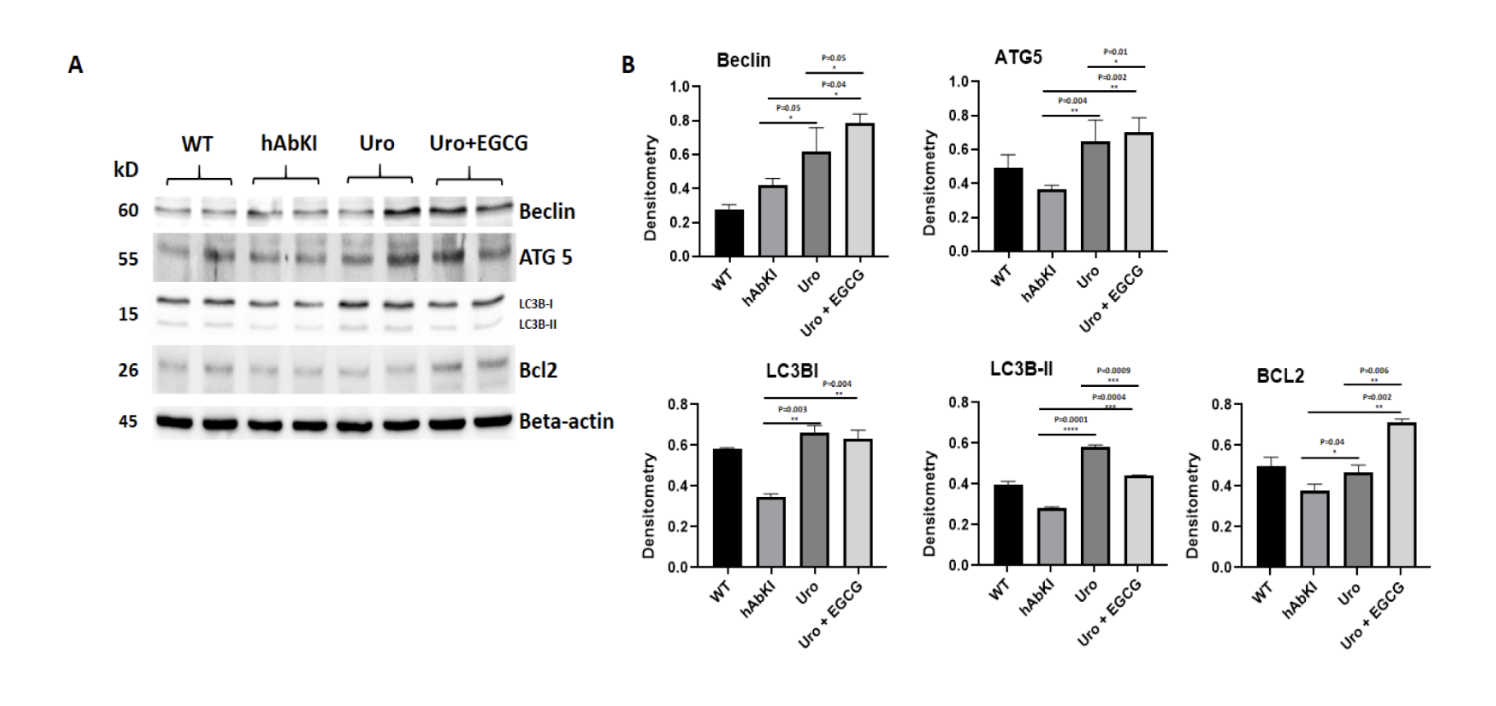
Figure 6: The broader autophagy machinery is comprehensively restored, more completely by the combination. ATG5, Beclin, BCL2, LC3B-I, and LC3B-II were all significantly elevated in treated mice, with the combination producing meaningfully higher levels of Beclin, ATG5, and BCL2 than urolithin A alone.
Mitochondrial Morphology and Function: Structural and Energetic Recovery
Electron microscopy provided a direct look at the mitochondria themselves, and the numbers confirm that the molecular improvements translated into structural ones. In the hippocampus, where Alzheimer's pathology concentrates its earliest and most damaging effects, urolithin A treated mice showed a significantly reduced number of fragmented mitochondria at p equals 0.0002, with the combination producing an even stronger reduction at p equals 0.0001. In the cerebral cortex, the pattern held at p equals 0.0004 for urolithin A and p equals 0.0002 for the combination. Mitochondrial length, which reflects the fusion-dominant network morphology associated with healthy energy production, increased significantly in both brain regions in both treatment groups, with the combination producing consistently stronger increases.
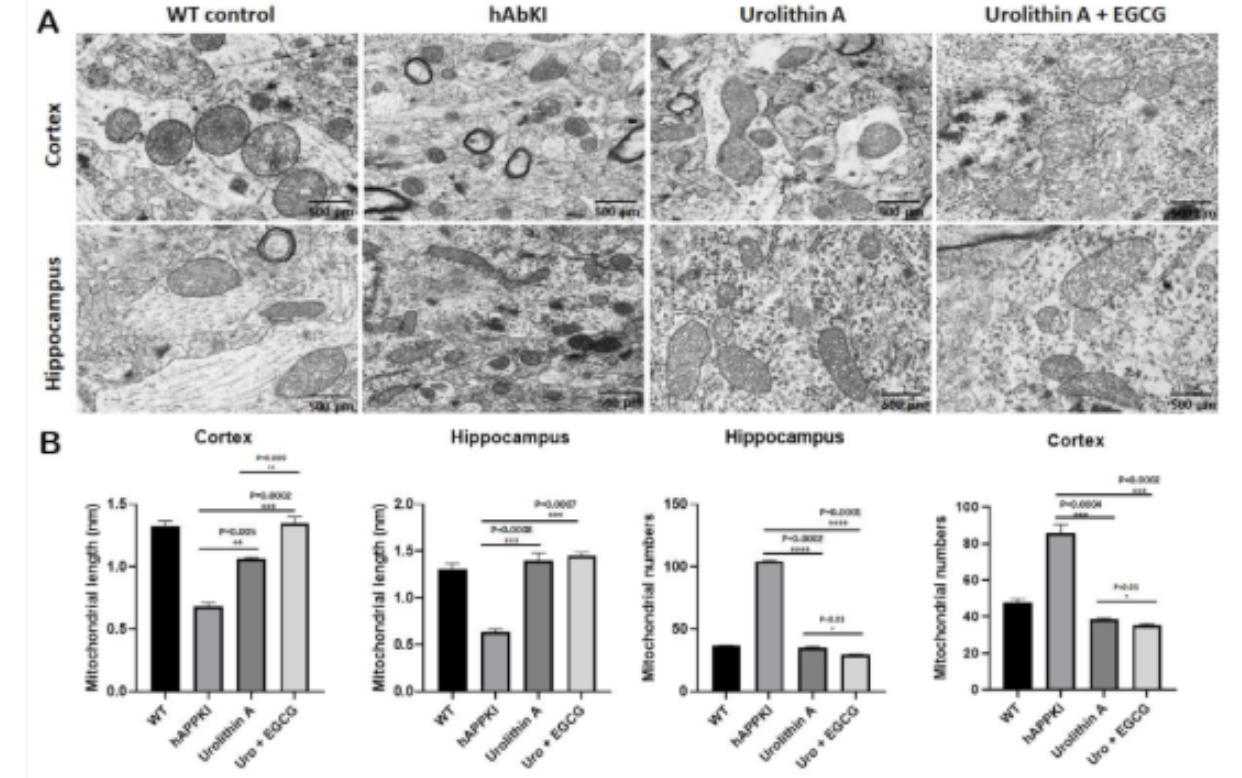
Figure 12: Mitochondrial fragmentation is visibly reduced and mitochondrial length restored by treatment. The shift from numerous small fragmented mitochondria in untreated animals toward fewer, longer, more connected structures in treated animals is directly visible. Quantitative analysis confirms stronger improvements with the combination across both brain regions.
The functional consequence of these structural improvements showed up in three direct biochemical measurements. Lipid peroxidation, the oxidative damage that dysfunctional mitochondria inflict on cellular membranes and one of the most sensitive indicators of mitochondrial stress, was significantly reduced in both treatment groups, with the combination producing a further significant reduction over urolithin A alone at p equals 0.03. Hydrogen peroxide, the reactive oxygen species generated when mitochondria are not running efficiently, was reduced in urolithin A treated mice at p equals 0.0008 and more strongly in the combination group at p equals 0.0004, with the combination versus urolithin A comparison reaching p equals 0.006. And mitochondrial ATP production, the ultimate output of a functioning mitochondrial network and the energy currency that neurons depend on for everything from firing to synaptic maintenance to amyloid clearance, increased significantly in both groups, reaching p equals 0.007 for urolithin A and p equals 0.0002 for the combination.
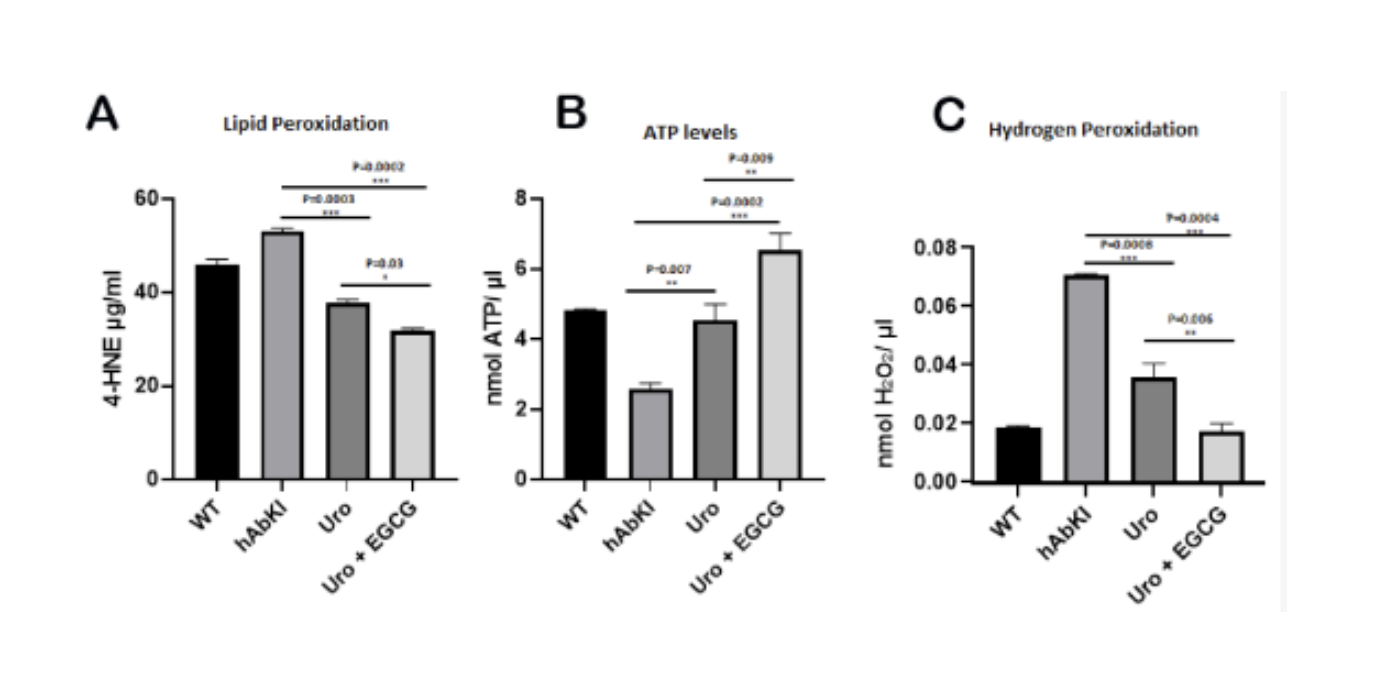
Figure 14: Mitochondrial function improves across all three direct measures. Lipid peroxidation fell, hydrogen peroxide production fell, and ATP output rose in both treatment groups. The combination produced significantly greater improvements than urolithin A alone across all three measures.
These three measurements together tell a coherent functional story. The mitochondria in treated mice were producing less oxidative stress, causing less oxidative damage, and generating more usable energy. The combination was doing all three more completely than urolithin A alone.
Synaptic Health and Dendritic Structure: Rebuilding the Infrastructure of Memory
The connection between mitochondrial recovery and cognitive improvement runs through the synapse, and the synaptic data shows that the molecular improvements were propagating downstream to the physical connections between neurons exactly as the metabolic hypothesis predicts.
The synaptic gene expression data was among the most striking in the study. In urolithin A treated mice, every synaptic gene measured showed significant upregulation: synaptophysin by 3.45-fold, PSD95 by 2.9-fold, synapsin 2 by 10.2-fold, synaptobrevin 2 by 6.6-fold, and neurogranin by 3.81-fold. These are the molecular building blocks of functional synaptic connections, and their restoration signals that the cellular environment was becoming hospitable to synaptic maintenance in a way that untreated hAbKI brains were not.
The combination produced consistently larger fold changes across every synaptic gene, but the most dramatic difference was in synapsin 1, which increased by just 2.25-fold with urolithin A but by 17.03-fold with the combination. Neurogranin reached 8.5-fold with the combination compared to 3.81-fold with urolithin A alone. These are not marginal differences. They suggest that EGCG, by reducing amyloid burden and neuroinflammation simultaneously, was creating a cellular environment in which the synaptic restoration that urolithin A's mitochondrial work was enabling could proceed much further than it could under the conditions urolithin A alone was able to establish.
The Golgi-Cox staining confirmed this at the structural level. Dendritic spine density increased significantly in hippocampal neurons of both treatment groups, but the combination's advantage was particularly apparent in the cortex, where urolithin A alone did not reach statistical significance for dendritic length while the combination reached p less than 0.0001. The physical infrastructure of memory and learning, the dendritic spines through which neurons receive signals from their neighbors, was being more completely restored by the combination than urolithin A could achieve independently.
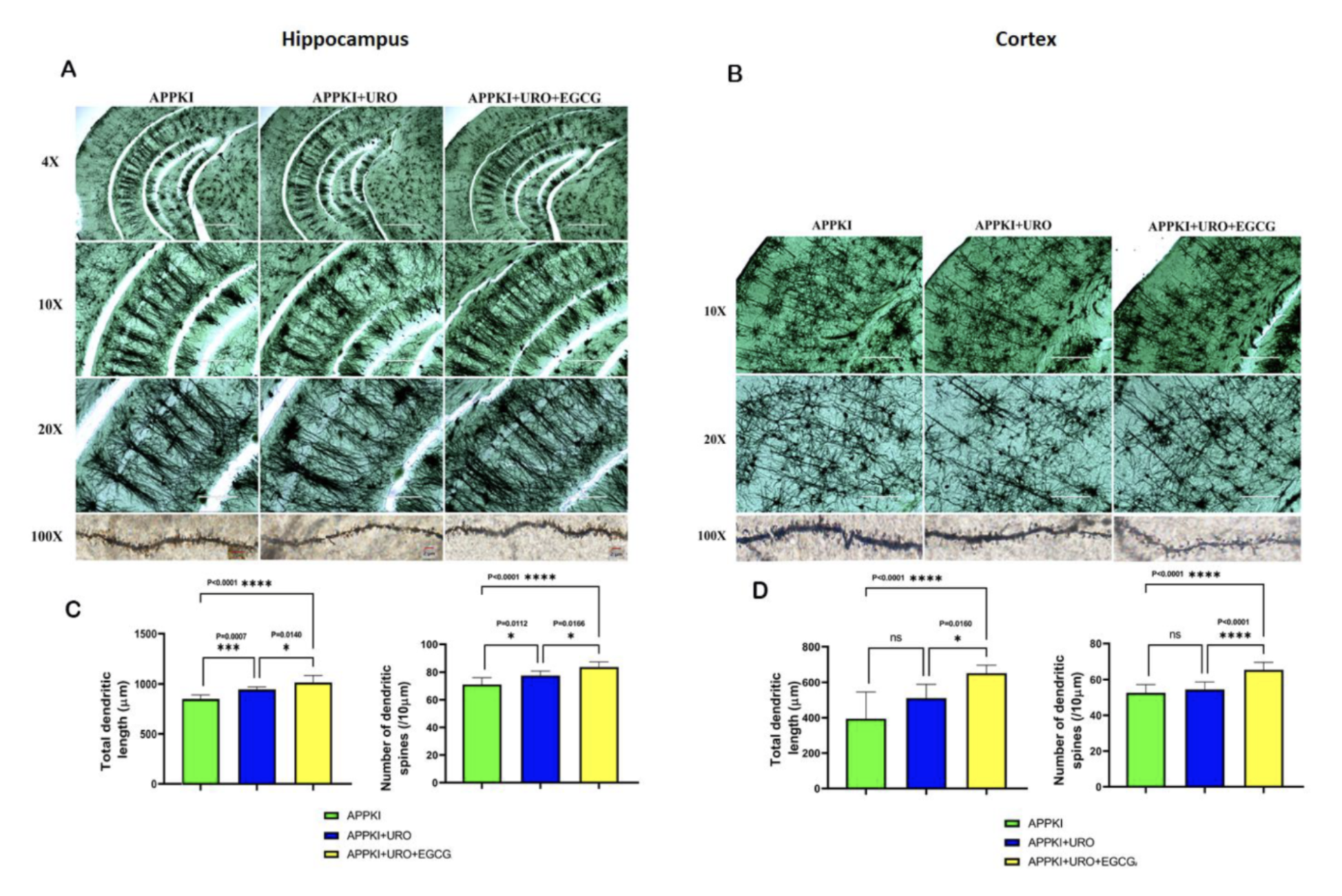
Figure 15: The physical infrastructure of memory is partially restored, more completely by the combination. Dendritic spine density increased significantly in both treatment groups. The combination produced stronger hippocampal restoration and a significant cortical effect that urolithin A alone did not achieve.
Neuroinflammation: Quieting the Amplifier
The neuroinflammatory findings added a dimension that sits at the intersection of the metabolic and the inflammatory hypotheses of Alzheimer's disease. Iba1, the microglial activation marker whose elevated levels signal that the brain's immune cells have shifted into the chronic inflammatory state that damages neurons alongside the pathological material they are activated against, was significantly reduced in urolithin A treated mice at p equals 0.002, and further reduced in the combination group at p equals 0.004. GFAP, reflecting astrocytic activation, was significantly reduced in both groups at p equals 0.02.
Crucially, the neuronal survival marker NeuN, which indicates the presence of intact functioning neurons, moved in the opposite direction, increasing significantly in urolithin A treated mice at p equals 0.02 and more strongly in the combination group at p equals 0.005. Neurons were being preserved. When the combination was compared directly to urolithin A, the combination produced significantly lower Iba1 at p equals 0.004, lower GFAP at p equals 0.02, and higher NeuN at p equals 0.006 across all three markers. The neuroinflammatory amplifier was being quieted more completely by the combination, and the neuronal population it had been threatening was more completely preserved.
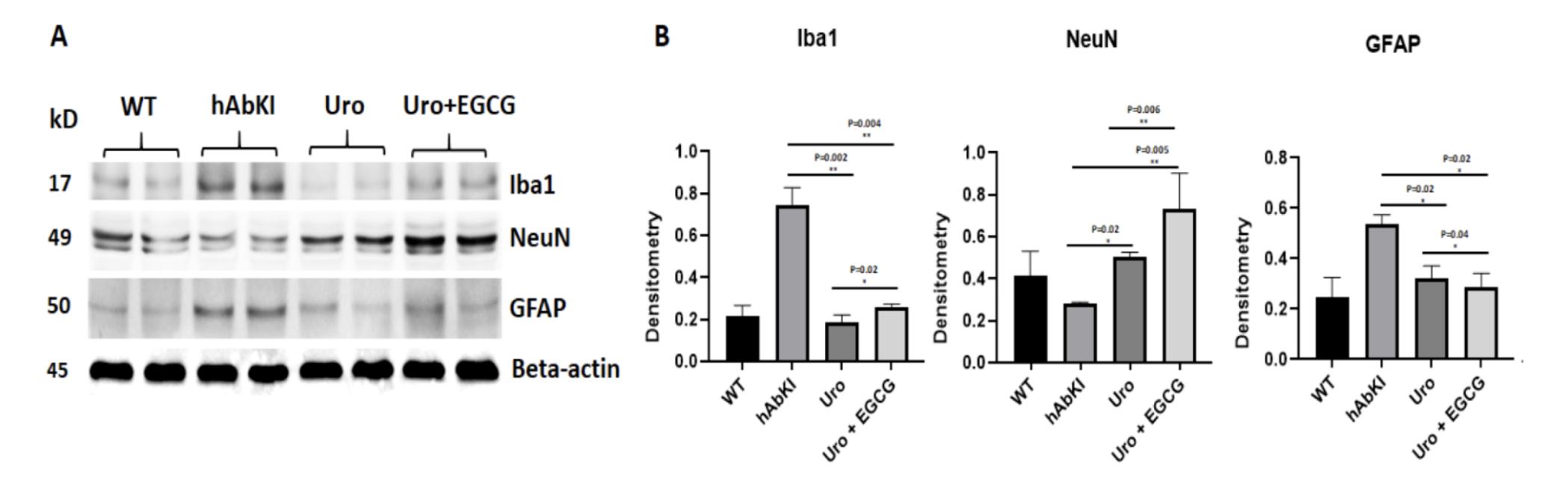
Figure 7: Neuroinflammation falls and neuronal survival improves simultaneously, more completely with the combination. Microglial marker Iba1 and astrocytic marker GFAP were significantly reduced while neuronal survival marker NeuN was significantly increased, with the combination producing stronger effects in both directions.
Amyloid-Beta: The Downstream Marker
The amyloid findings close the loop on the metabolic hypothesis in a way that is worth stating carefully. Soluble amyloid-beta 40 and amyloid-beta 42 were both progressively decreased in treated hAbKI mice, with the combination producing the larger reduction across both species.
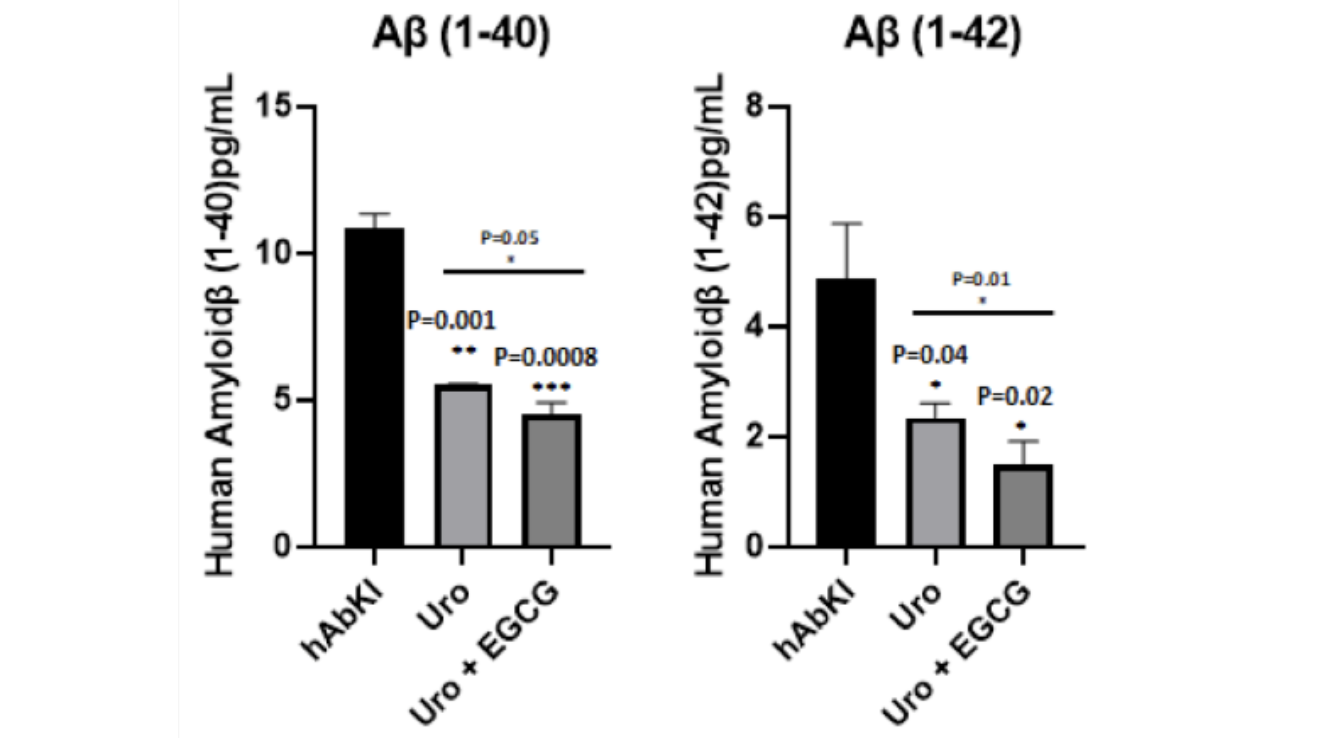
Figure 16: Amyloid-beta levels decline with treatment, more substantially with the combination. Both Aβ40 and Aβ42 were reduced in treated mice relative to untreated seven-month-old controls, with the combination producing the larger reduction across both species.
Within the metabolic hypothesis framing, amyloid accumulation reflects not simply increased amyloid production but impaired amyloid clearance driven by energy failure. An intervention that restores mitochondrial function should therefore improve amyloid clearance and reduce amyloid burden even without directly targeting the amyloid biology. That is precisely what the data show. The intervention targeted the upstream mitochondrial failure. Energy production improved. Oxidative stress fell. The cellular machinery responsible for amyloid processing recovered. And amyloid levels declined as a result. The combination, by additionally targeting amyloid aggregation through EGCG's direct anti-amyloid mechanisms, produced a larger reduction than the mitochondrial intervention alone could achieve.
This sequence, from mitochondrial restoration through energy recovery to amyloid clearance, is the metabolic hypothesis rendered as a dataset. Each step in the predicted causal chain showed movement in the expected direction. The intervention targeting the upstream metabolic failure produced downstream effects on amyloid that an amyloid-first intervention has consistently failed to produce in the other direction. That asymmetry is not proof of the hypothesis. But it is the result the hypothesis predicts, and its consistent appearance across both treatment groups, stronger in the combination, is the kind of evidence that makes the metabolic framing of Alzheimer's disease increasingly difficult to dismiss.
Why the Combination Works Better: The Mechanistic Logic
The most practically significant finding in this study is not that urolithin A worked. The prior cell culture work from this research group had already established that with considerable consistency. The most significant finding is that adding EGCG to urolithin A produced systematically stronger effects across every outcome measured, often by margins that are difficult to attribute to simple additive effects of two compounds with overlapping mechanisms. Understanding why requires stepping back from the data and examining how the two compounds' mechanisms relate to each other within the cascade the metabolic hypothesis describes.
Urolithin A's primary contribution is to the cellular machinery that manages mitochondrial quality. It activates PINK1 and Parkin, shifting the balance of the mitochondrial network toward fusion over fission, stimulating the biogenesis program that replaces cleared mitochondria with healthy ones, and restoring the autophagosomal machinery that physically executes the clearance process. In the context of the metabolic hypothesis, urolithin A is working from within the mitochondrial system outward, restoring the energy infrastructure that amyloid clearance and synaptic maintenance depend on, and creating the cellular conditions under which downstream recovery can begin.
EGCG is working from a different entry point entirely. Its primary contributions are upstream of the mitochondrial system in one direction and downstream of it in another. On the upstream side, EGCG directly targets amyloid-beta aggregation, redirecting amyloid peptides away from the fibrillar forms that interact with mitochondrial proteins and disrupt the PINK1-Parkin pathway, and converting existing toxic fibrils into less damaging assemblies. By reducing the amyloid burden that is actively undermining the mitochondrial quality control system that urolithin A is trying to restore, EGCG removes one of the primary forces working against urolithin A's mechanism. On the downstream side, EGCG suppresses microglial activation and the neuroinflammatory environment that mitochondrial dysfunction produces and that, once established, amplifies cellular damage independently of whatever triggered it.
The practical implication of this mechanistic complementarity is that urolithin A and EGCG are not doing the same thing twice. They are each doing something the other cannot, and those contributions reinforce each other in ways that produce effects larger than either could generate independently. Urolithin A restores the cellular maintenance system that amyloid has broken. EGCG reduces the amyloid burden doing the breaking while simultaneously quieting the inflammatory response that the broken maintenance system has generated. When both compounds are present simultaneously, the quality control restoration that urolithin A drives is happening in an environment where the primary forces opposing it have been reduced by EGCG, and the anti-amyloid and anti-inflammatory effects of EGCG are being amplified by the improved cellular energy environment that urolithin A's mitochondrial restoration is creating.
The synaptic gene data captures this synergy most vividly. Synapsin 1, one of the key proteins governing synaptic vesicle function, increased by just 2.25-fold with urolithin A but by 17.03-fold with the combination. That sevenfold amplification from adding EGCG to a urolithin A protocol is not the expected result of simply combining two moderately effective compounds. It suggests that urolithin A was creating the cellular conditions for synaptic recovery that the inflammatory and amyloid burden in untreated mice was preventing, and that EGCG was removing that burden sufficiently to allow the synaptic recovery program to proceed to a degree that urolithin A's mitochondrial restoration alone could not unlock.
The mitophagy formation data from transmission electron microscopy adds a further dimension to this interpretation. More mitophagosomal formations were visible in the combination group than in the urolithin A group, suggesting that EGCG was not simply adding its own effects alongside urolithin A's but was amplifying the mitophagy process itself, possibly by reducing the amyloid-tau burden that directly interferes with the PINK1-Parkin pathway and thereby allowing urolithin A's mitophagy enhancement to operate more efficiently in the reduced-burden environment EGCG was creating.
Whether this amounts to true synergy, in the pharmacological sense of a combined effect that exceeds what additive effects of the two compounds' independent mechanisms would predict, is a question the current study was not designed to answer formally. Establishing that would require dose-response experiments comparing the combination to each compound alone at multiple concentrations, which remains an important direction for future work. What the current data establish is that the combination is consistently and substantially better than urolithin A alone across every outcome measured, that the mechanistic logic for why is coherent and grounded in the distinct biological profiles of the two compounds, and that the magnitude of certain effects, particularly in the synaptic gene data, suggests the combination is producing something more than the arithmetic sum of its parts.
What Needs to Be Answered Next
The study points clearly toward several directions that would meaningfully extend its findings. Dose-response experiments with oral administration of urolithin A and the combination would establish whether the effects demonstrated here can be replicated under conditions that more closely approximate clinical use. Studies beginning treatment after established pathology would address the therapeutically most important question the current design leaves open. Larger cohorts including both sexes, given evidence that sex influences Alzheimer's disease progression and response to interventions in ways the current study cannot address, would provide more robust estimates of effect sizes and their variability. And human studies examining the effects of urolithin A supplementation on mitochondrial function in aging brains, ideally using biomarkers that can be measured non-invasively, would begin the necessary work of establishing whether the biology demonstrated here operates similarly in the species the intervention is ultimately intended to help.
The combination of urolithin A and EGCG has earned the right to be studied further. Whether it earns the right to be used clinically is a question that requires the kind of rigorous human evidence that this preclinical study, however carefully designed, cannot provide. The distance between these findings and a validated Alzheimer's intervention remains substantial. The direction they point is worth following.
Conclusion: What This Study Tells Us About Alzheimer's Disease and How to Fight It
The story this study tells is ultimately not just about urolithin A or EGCG. It is about what happens when you target Alzheimer's disease at its metabolic foundation rather than at its most visible downstream consequence.
For more than two decades, the dominant approach to Alzheimer's drug development has been to target amyloid directly, clearing plaques, blocking their formation, or neutralizing their toxic effects. The clinical record of that approach is, with limited exceptions, a record of failure. Drugs that reduced amyloid burden in the brain did not reliably produce cognitive benefit. The field has been forced to ask why, and the metabolic hypothesis offers one of the more compelling answers: because amyloid accumulation is not the initiating event but a downstream consequence of a brain that has lost its metabolic footing. Clearing the consequence without addressing the cause is unlikely to produce durable benefit, which is precisely what the trial record reflects.
The hAbKI model used in this study was designed with this reframe in mind. By producing human amyloid-beta under normal physiological regulatory control rather than through artificial overexpression of familial mutations, it creates a biological context where mitochondrial dysfunction and defective mitophagy are not simply consequences of catastrophic genetic overexpression but emerge through the same gradual metabolic deterioration that characterizes late-onset human Alzheimer's disease. Testing urolithin A in this model is therefore testing the metabolic hypothesis more directly than prior preclinical Alzheimer's research has generally allowed.
What the data show across seven outcome domains is consistent with the hypothesis in a way that is difficult to dismiss even accounting for the translation uncertainties that every mouse study carries. Restoring mitochondrial quality control through urolithin A's mitophagy enhancement produced improvements not just in mitochondrial biology but in synaptic health, neuroinflammation, and amyloid levels simultaneously. The cascade that the metabolic hypothesis places at the center of Alzheimer's pathology showed evidence of being interrupted at its origin rather than managed at its endpoints. And the combination with EGCG, by addressing the amyloid burden and inflammatory environment that mitochondrial dysfunction generates while urolithin A restored the mitochondrial machinery that amyloid accumulation had impaired, produced a more complete interruption across every level of the cascade than either compound could achieve working from a single mechanistic entry point.
None of this constitutes proof that urolithin A or the combination will work in humans. The distance between a carefully designed mouse study and a validated human intervention is real and should not be minimized. The delivery problem, the timing question, the group size limitations, and the fundamental challenge of translating mouse biology to human Alzheimer's disease across decades of accumulated complexity are all genuine constraints on how confidently the findings can be extrapolated. What the study provides is not a clinical answer but a scientifically credible direction, grounded in a more realistic model of the disease than most preclinical research has used, and pointing toward a mechanism that the weight of evidence increasingly suggests is central to how Alzheimer's disease actually develops.
The broader implication reaches beyond any single compound or combination. If defective mitophagy is genuinely upstream of amyloid accumulation in late-onset Alzheimer's disease, as the metabolic hypothesis proposes and as these findings are consistent with, then the therapeutic strategy for Alzheimer's may need to be reoriented from targeting what the disease produces to restoring what the disease destroys. Amyloid is not nothing. But it may be less the cause of Alzheimer's disease than the signature of a brain that has lost the metabolic capacity to clear it. A compound that restores that capacity is not treating a symptom. It is addressing the failure that allows the symptoms to accumulate in the first place.
Urolithin A and EGCG are not going to cure Alzheimer's disease. But the evidence from this study suggests they are engaging the right biology, in a more realistic model than the field has typically tested, with effects that propagate across the disease cascade in the direction the metabolic hypothesis predicts. That is a meaningful result. It is also, in the context of a disease that has resisted every pharmacological attempt to alter its course, an encouraging one.
Related studies