Evaluating the Evidence: Which mTOR Interventions Actually Work in Living Humans
mTOR is the molecular switch at the center of biological aging. When chronically active, mTOR suppresses the cellular maintenance programs that keep tissue healthy. Autophagy switches off, damaged proteins accumulate, dysfunctional mitochondria linger, and the inflammatory burden that defines biological aging builds. Every major hallmark of aging sits downstream of this single regulatory node.
Rapamycin is the most potent and reproducible mTOR inhibitor identified to date. It extends lifespan across an extraordinary range of organisms, including a 9 to 14% increase in genetically diverse mice even when treatment was initiated late in life. No other pharmacological intervention has demonstrated comparable consistency across model organisms.
Low-dose intermittent rapamycin improves immune function in older humans. In a landmark 2014 randomized trial, adults over 65 receiving weekly mTOR inhibition demonstrated up to a 20% greater antibody response to influenza vaccination alongside a roughly 30% reduction in exhausted immune cells that can no longer respond effectively to new threats.
The PEARL trial demonstrated measurable body composition benefits in healthy older adults. Over 48 weeks, women receiving 10 mg weekly rapamycin gained an average of 4.5% in lean muscle mass while men experienced a 1.4% increase in bone mineral content at a life stage where the placebo group was losing bone.
A 2025 ME/CFS trial provided direct human evidence that restoring autophagy through mTOR inhibition translates into clinical benefit. BECLIN-1, a key marker of cellular cleanup activity, rose by roughly 40 to 50% over 90 days while the molecular brake on autophagy fell by more than two-fold. Patients showing the greatest restoration of autophagy signaling reported the largest improvements in fatigue and activity tolerance.
Natural compounds engage the mTOR pathway indirectly and with substantially lower potency than rapamycin. Resveratrol activates AMPK to suppress mTOR from upstream. Curcumin quiets the signaling cascade driving mTOR activation while reducing inflammatory tone. EGCG directly engages the autophagy ignition machinery. Berberine mimics caloric restriction through mitochondrial energy interference. Each mechanism is coherent, but bioavailability constraints limit real-world impact.
The gap between mechanistic plausibility and real-world impact is the central challenge for natural compounds. Resveratrol suppresses mTOR at concentrations far exceeding what oral supplementation achieves in circulation. Curcumin demonstrates poor systemic absorption even at doses as high as 12 grams. The cellular biology is compelling. The delivery problem is real.
Combining rapamycin with natural compounds may produce synergistic effects that neither achieves alone. A 2015 preclinical study found that rapamycin combined with resveratrol reduced a key compensatory survival signal by approximately 45% compared to rapamycin alone, while cellular proliferation markers fell by roughly 50% and programmed cell death in abnormal cells increased by nearly threefold.
Structured fasting can engage the mTOR-autophagy axis through the body's own biology. A 2025 randomized trial found that a five-day fasting-mimicking diet produced measurable acceleration of autophagic flux in living humans, with the effect persisting two days after normal eating resumed.
The human evidence base is growing but definitive proof of lifespan extension in humans remains absent. Low-dose rapamycin has demonstrated improvements in immune function, body composition, and cellular repair signaling across multiple trials. What it has not established is whether these benefits translate into longer lives. That question requires larger and longer trials than have yet been completed.
For decades, the pursuit of longevity was largely confined to speculation, often characterized as a search for a mythical fountain of youth. Today, however, that pursuit has moved into the realm of molecular biology. We are no longer asking whether we can slow the clock, but which biological mechanisms we must target to meaningfully influence it. This shift has brought attention to a central regulatory pathway within cells: the mechanistic target of rapamycin, a protein kinase that helps govern how biological wear accumulates over time. And of all the questions modern aging biology is now positioned to answer, the question of how to modulate this pathway, safely, precisely, and sustainably across a human lifespan, may be the most consequential.
What is mTOR? The Switch at the Center of Cellular Aging
Every cell in the body faces a fundamental resource allocation problem. It can invest its energy in growing, replicating, and building new proteins. Or it can invest in maintaining what it already has, clearing out damaged components, repairing stress-induced wear, and shoring up its defenses against future insults. It cannot do both at full capacity simultaneously.
The protein that sits at the center of this decision is called mTOR, the mechanistic target of rapamycin. Discovered in the late 1990s, mTOR functions as the cell's master growth regulator, a molecular switchboard that continuously reads signals from the surrounding environment and adjusts cellular behavior accordingly [1]. It does this through a process called phosphorylation, the addition of a charged phosphate group to other proteins, which acts like a molecular on/off switch, activating or silencing downstream processes depending on what the cell needs at any given moment.
The logic of mTOR's signaling is elegantly simple. When nutrients are abundant and growth factors like insulin are circulating, mTOR reads the environment as favorable and pushes the cell toward expansion: synthesizing proteins, building new structures, and preparing to divide. When nutrients are scarce, when stress signals arrive, or when the cell's energy reserves run low, mTOR activity falls, and the cell pivots toward a different set of priorities. It begins conserving resources, activating repair pathways, and clearing out the molecular debris that accumulates during normal operation.
In the context of a young, healthy organism, this system works beautifully. Growth when conditions are good. Maintenance when they are not. The problem emerges over decades.
As we age, mTOR tends toward chronic activation, driven partly by the persistent nutrient abundance of modern diets and partly by the hormonal and metabolic changes that accumulate with age. In this chronically active state, the cell's maintenance programs are perpetually suppressed. The most consequential of these is autophagy, the cellular recycling system that identifies and breaks down damaged proteins and dysfunctional organelles. Think of autophagy as the cell's waste management service. When mTOR keeps it switched off, the waste accumulates. Damaged proteins aggregate. Dysfunctional mitochondria linger rather than being cleared and replaced. Oxidative stress rises as cellular quality control deteriorates.
The consequences ripple outward. Mitochondrial efficiency declines, reducing the cell's capacity to produce energy. A state of chronic low-grade inflammation, increasingly referred to in aging research as inflammaging, takes hold as the accumulated cellular debris triggers immune responses. And cells that should either repair themselves or undergo programmed death instead persist in a dysfunctional state called senescence, secreting inflammatory signals that gradually impair the function of surrounding tissue [3].
Each of these processes, suppressed autophagy, mitochondrial decline, inflammaging, and cellular senescence, is now recognized as a hallmark of biological aging. And mTOR sits upstream of all of them. That is what makes it such a compelling target for longevity research, and what makes the question of how to modulate it one of the most actively investigated problems in modern aging biology [3,4].
Rapamycin: Targeting mTOR to Reprogram Cellular Aging
The story of rapamycin begins not in a laboratory but in one of the most remote places on earth.
In the 1960s, a scientific expedition to Rapa Nui, the Chilean territory better known as Easter Island, collected soil samples from the island's volcanic terrain. Among the microorganisms found in those samples was a bacterium called Streptomyces hygroscopicus, which produced a compound that would eventually take the island's name: rapamycin. Initially investigated as an antifungal agent, its most significant properties turned out to lie elsewhere entirely. Decades of subsequent research revealed it to be one of the most potent and reproducible pharmacological interventions for lifespan extension ever identified, extending healthy lifespan across an extraordinary range of organisms, from single-celled yeast to fruit flies, worms, and mammals [5].
The mechanism is direct. Rapamycin binds to an intracellular protein called FKBP12, and the resulting complex latches onto mTOR and inhibits its activity. Where chronic mTOR activation had been pushing cells toward growth at the expense of maintenance, rapamycin tips that balance in the other direction. Cellular resources shift away from energy-intensive protein synthesis and replication and toward the repair and stress-resistance programs that aging progressively suppresses. Autophagy, the cellular waste management system introduced in the previous section, is reactivated. Damaged proteins and dysfunctional organelles are cleared. Mitochondrial function stabilizes. The inflammatory signaling that accumulates with chronic mTOR activity begins to quiet.
In this sense, rapamycin does not simply slow a biological process. It induces an adaptive shift in cellular priorities, one that the cell's own machinery is capable of executing but that chronic mTOR activation had been preventing. The question that has consumed researchers for the past decade is whether that shift, so consistently beneficial in model organisms, translates into meaningful outcomes in humans.
Beyond Rapamycin: How Diet, Energy, and Lifestyle Regulate mTOR
Rapamycin is not the only way to shift mTOR's balance toward maintenance and repair. The pathway did not evolve to wait for a compound from Easter Island soil. It evolved to respond to the metabolic reality of the organism moment to moment, and the signals it reads are ones that diet, exercise, and lifestyle generate continuously.
At its core, mTOR is an integrator. It sits at a cellular crossroads where signals from multiple directions converge, and it adjusts cellular behavior based on the combined picture they paint. When insulin and its close relative IGF-1 are circulating at high levels, as they are after a meal rich in carbohydrates and protein, mTOR reads the environment as nutrient-abundant and activates accordingly. When calories are restricted, when a meal is delayed, or when the body is deep into a fasting state, those signals quiet down and mTOR activity falls with them.
A second input comes from a protein called AMPK, or AMP-activated protein kinase, which functions as the cell's dedicated energy gauge. When cellular energy reserves run low, as they do during intense exercise or prolonged fasting, AMPK is activated and sends an inhibitory signal to mTOR, effectively telling the cell that now is not the time for growth. Conserve, repair, and wait. This is one of the primary reasons that fasting and exercise are associated with many of the same cellular benefits as direct mTOR inhibition: they engage the same upstream regulatory machinery through the body's own physiology.
This regulatory architecture has generated significant interest in natural compounds that might partially mimic these metabolic signals. Resveratrol, found in grape skins and berries, activates AMPK and thereby indirectly suppresses mTOR. Curcumin, the active compound in turmeric, modulates upstream signaling pathways that feed into mTOR activity. Others, including epigallocatechin gallate from green tea and withaferin A from ashwagandha, engage overlapping mechanisms. The appeal is obvious: if the body already uses these metabolic signals to regulate mTOR, perhaps compounds that amplify those signals could produce meaningful longevity benefits through diet and supplementation alone.
The honest answer requires looking carefully at what the research actually shows. The sections that follow examine the human evidence for rapamycin first, then the natural compounds, before turning to the question of how they compare and whether combining them might offer something neither achieves alone.
Rapamycin: The Human Evidence Base
Model organisms have made the case for mTOR inhibition compellingly. Rapamycin extends lifespan in yeast, worms, flies, and mice, often dramatically, and across a range of genetic backgrounds and ages of initiation. But the history of longevity research is littered with interventions that worked beautifully in animals and failed to translate to humans. The biology is more complex, the timescales are longer, and the endpoints are harder to measure. The question that ultimately matters is whether carefully dosed mTOR inhibition produces meaningful benefits in people.
Over the past decade, that question has begun to receive answers.
Reversing Immune Aging: mTOR and Immunosenescence
One of the most well-documented consequences of aging is a gradual deterioration of immune function known as immunosenescence. The immune system becomes less efficient at recognizing new threats, slower to mount responses, and increasingly populated by cells that have been activated so many times they have lost their effectiveness. The practical consequences are familiar: older adults are more susceptible to infection, recover more slowly when they do get sick, and respond less robustly to vaccines.
A landmark 2014 study by Mannick and colleagues asked whether low-dose mTOR inhibition could reverse some of this decline [6]. The trial enrolled more than 200 adults over the age of 65 and assigned them to one of four groups: a low daily dose of RAD001, an mTOR inhibitor closely related to rapamycin, at 0.5 mg, two intermittent weekly dosing regimens at 5 mg or 20 mg, or placebo. After six weeks of treatment followed by a two-week washout period, all participants received a standard influenza vaccine, and immune response was assessed by measuring antibody titers, the concentration of antibodies generated against the vaccine's target strains. Higher titers indicate a more robust and effective immune response.
The results were notable. Participants who received RAD001 demonstrated up to a 20% greater antibody response compared to those on placebo, a difference the study defined as clinically meaningful. The group receiving 20 mg once weekly showed the most consistent improvements, particularly against the H3N2 influenza strain, where antibody responses crossed the threshold for clinical significance. Lower doses produced more modest and variable benefits depending on the strain.
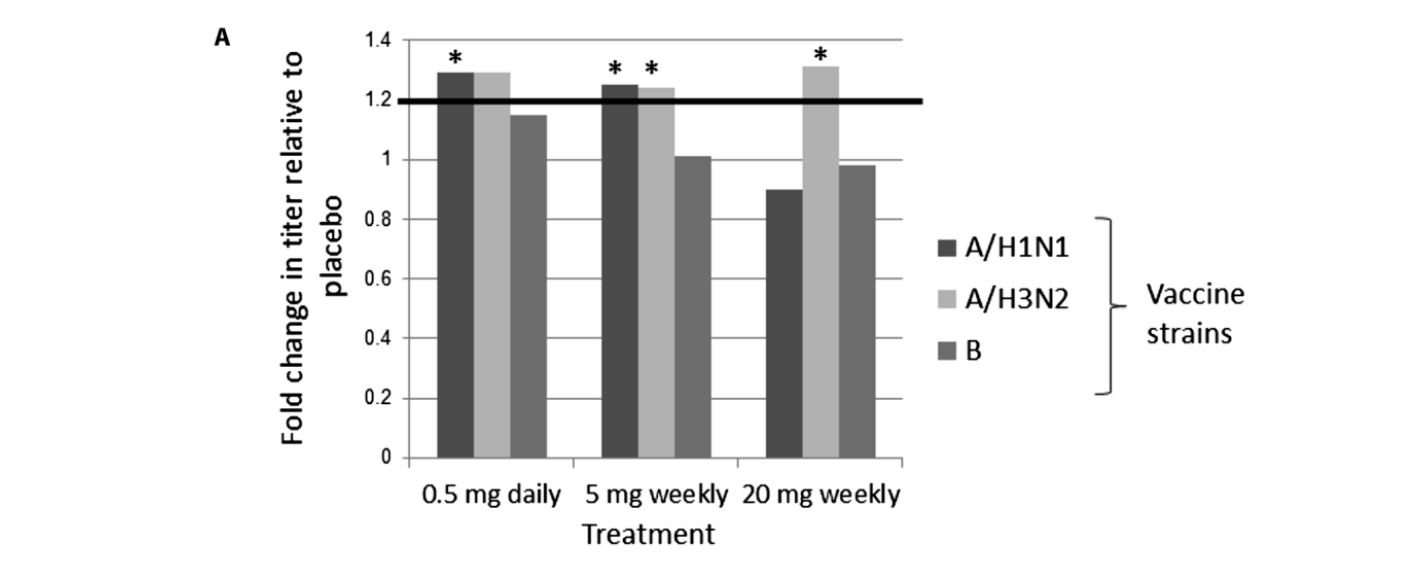
Figure 1: Increase in antibody levels against three influenza strains (A/H1N1, A/H3N2, and B) in participants receiving different doses of RAD001, shown relative to the placebo group. Reprinted from. “mTOR inhibition improves immune function in the elderly,” by J.B. Mannick et al., 2014. Science translational medicine, Volume 6 Issue 268.
The PEARL Trial (2025)
The Mannick study established that mTOR inhibition could improve immune function in older adults. What it could not answer was whether those benefits extended beyond the immune system, and whether they persisted over longer timeframes. The PEARL trial, or Participatory Evaluation of Aging with Rapamycin for Longevity, was designed to push into that territory.
The trial followed 115 healthy adults between the ages of 50 and 85 over 48 weeks, making it the largest and longest randomized controlled trial of rapamycin in healthy older adults conducted to date [7]. Rather than focusing on immune markers alone, it examined whether weekly low-dose rapamycin could influence the physical architecture of aging itself: the loss of muscle mass, the decline in bone density, and the accumulation of visceral fat that collectively define the trajectory toward frailty.
The results supported a lower-dose, intermittent approach. At once-weekly dosing, rapamycin appeared to function less as a blunt pharmacological intervention and more as a modulatory signal, nudging underlying physiology in a direction that the aging process had been pushing against. One of the more intriguing findings was that the pattern of benefit differed meaningfully between men and women, suggesting that the biology of mTOR modulation interacts with sex-specific hormonal and metabolic context in ways that future trials will need to account for.
Moving the Needle on Muscle and Bone
Among adults over 60, the loss of lean muscle mass and bone mineral density is not a dramatic event. It is a slow, largely invisible deterioration that accumulates across years, becoming legible only when its consequences arrive: a fall, a fracture, a loss of the functional independence that had previously been taken for granted. The PEARL trial asked whether rapamycin could interrupt that trajectory.
The answer, at least at the 10 mg weekly dose, was yes, partially and in ways that differed by sex.
Women receiving the 10 mg weekly dose demonstrated an average gain of approximately 4.5% in lean muscle mass over the 48-week period, a meaningful change in a population where maintaining existing muscle is typically considered a success [7]. The range of individual responses was wide, with one participant gaining 19% lean mass over the course of the study, a figure that underscores both the potential and the variability of this approach.
Men showed a different pattern of benefit. Rather than muscle gains, the most pronounced improvements in the male participants appeared in bone health. Those receiving the 10 mg weekly dose experienced a 1.4% increase in bone mineral content over 48 weeks, at a life stage where the placebo group was losing bone [7]. Given that fracture risk is one of the strongest predictors of functional decline and mortality in older adults, even modest improvements in bone mineral content carry implications that extend well beyond the laboratory.
These findings are further illustrated in the figures below:
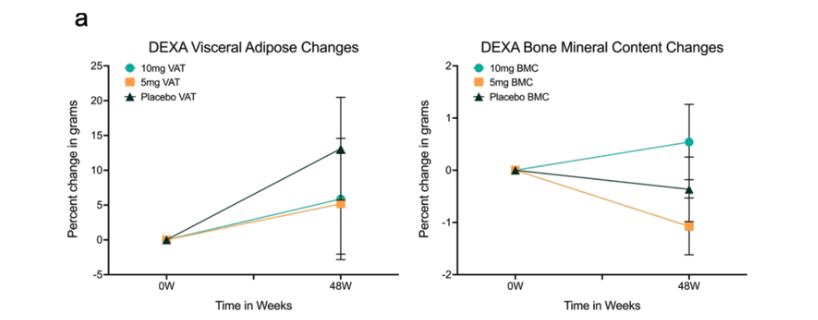
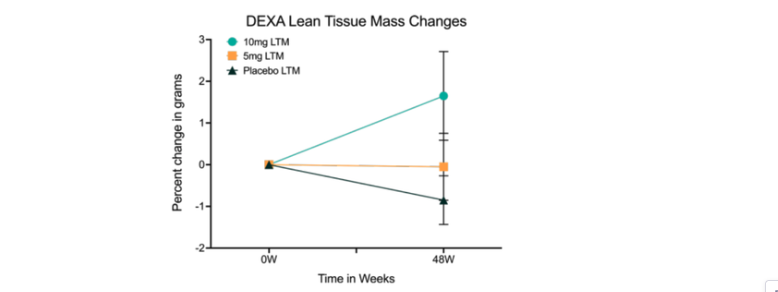
Figure 2. Body composition changes associated with low-dose weekly rapamycin. Percent change in visceral fat, bone mineral content, and lean tissue mass over 48 weeks for 10mg, 5mg, and placebo cohorts.
This set of three graphs illustrates how participants’ body composition changed over the 48-week study. Each graph uses baseline (0 weeks) as the reference point, with the vertical axis representing percentage change in grams. This allows changes to be interpreted relative to each participant’s starting point.
DEXA Visceral Adipose (Belly Fat):
The first graph shows visceral fat. The placebo group (dark triangles) exhibited a steady increase over time, while both the 5 mg and 10 mg rapamycin groups showed a much flatter trajectory, suggesting attenuation of age-related fat accumulation. [7]
DEXA Bone Mineral Content (Bone Strength):
The middle graph highlights bone health. The 10 mg group (teal circles) demonstrates a clear upward trend, indicating increased bone mineral content. In contrast, both the 5 mg and placebo groups experienced declines, suggesting a potential protective effect of the higher dose on skeletal integrity.
DEXA Lean Tissue Mass (Muscle):
The final graph tracks lean tissue mass. While the placebo and 5 mg groups remained stable or declined slightly, the 10 mg group showed a distinct increase, consistent with preservation or gain of muscle mass over time.
These changes in body composition underscore why this trial was previously highlighted as a notable development in longevity research.
From Biology to Daily Life: Functional and Healthspan Outcomes
Beyond physical changes, the trial also evaluated healthspan, defined as the number of years lived in good health rather than simply total lifespan. To assess this, researchers used the WOMAC index, a validated clinical tool that measures pain, stiffness, and physical function in individuals with osteoarthritis.
Osteoarthritis is characterized by the gradual breakdown of cartilage that cushions the joints, leading to pain, stiffness, and reduced mobility. In this study, women receiving rapamycin reported significant reductions in joint pain along with improvements in functional capacity and daily activity. These findings suggest that the benefits extended beyond structural changes, reflecting meaningful improvements in quality of life and physical independence. The results are further illustrated in Figure 3 below.
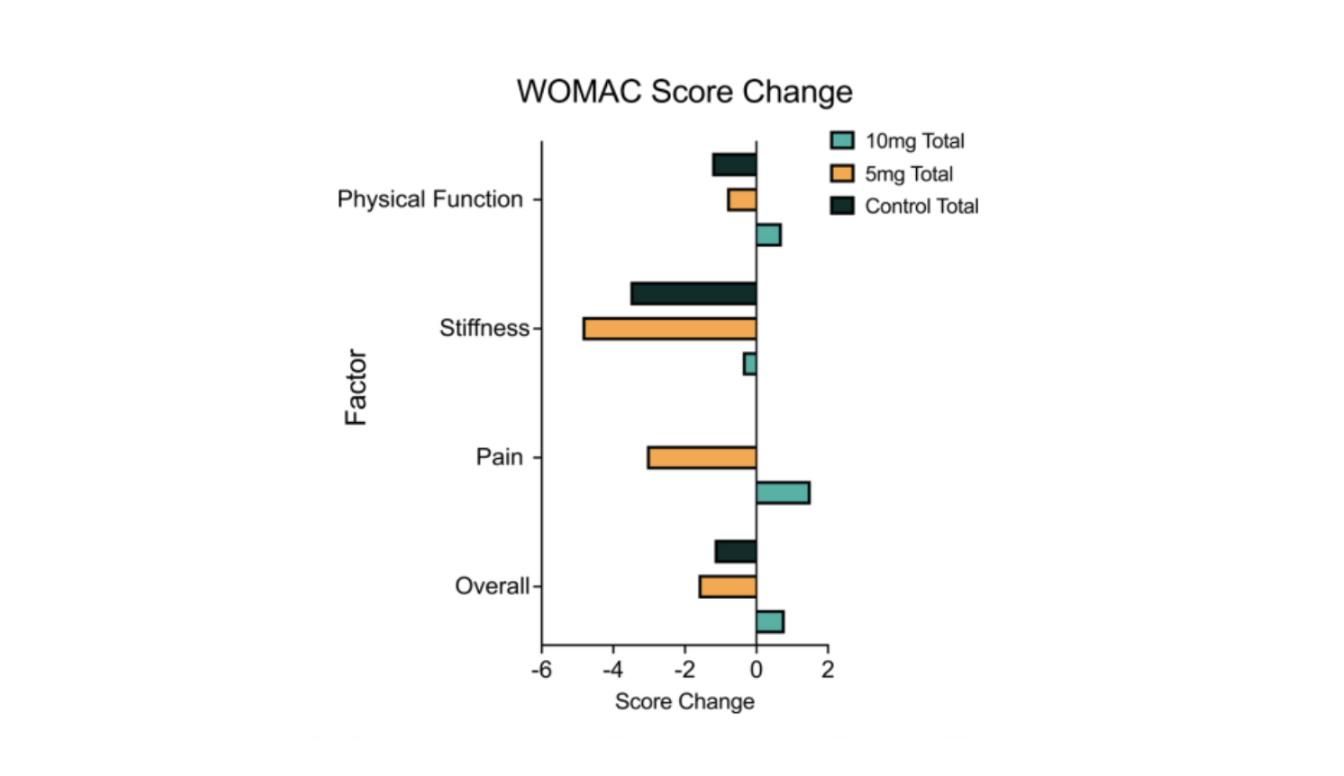
Figure 3. Osteoarthritis WOMAC score changes with rapamycin. The horizontal bar chart shifts focus from structural changes to patient-reported experience. The x-axis represents change in WOMAC score. In this scale, negative values (bars extending left) indicate symptom improvement, while positive values reflect worsening.
- Stiffness and Pain: Stiffness and Pain: The 5 mg group (orange bars) shows the most pronounced improvements, particularly in stiffness and pain, indicating reduced joint discomfort and improved mobility relative to baseline.
- Physical Function and Overall Score: The overall score, which captures cumulative impact, further highlights this pattern. While the 10 mg group (teal) remained near baseline or showed modest variation, the 5 mg group demonstrated more consistent improvements across functional domains.
These findings reinforce the concept of healthspan. They show not only measurable changes in body composition (Figure 1), but meaningful improvements in how individuals feel and function in daily life.
Overall, this trial helps bridge the gap between theoretical dosing strategies and real-world outcomes discussed more in depth in our previous article, Rapamycin Dosing for Longevity. It suggests that moderate mTOR inhibition can support maintenance and repair processes that preserve both structural integrity and functional capacity [8].
Rapamycin and ME/CFS: What Chronic Fatigue Syndrome Reveals About Autophagy
The clinical studies examined so far have measured rapamycin's effects on the body composition and immune function of healthy older adults. A 2025 trial took a different approach, applying the same biological logic to a population defined not by age but by a specific failure of cellular recovery: patients with myalgic encephalomyelitis and chronic fatigue syndrome, better known as ME/CFS.
ME/CFS is one of the most poorly understood chronic illnesses in modern medicine. Its defining feature is post-exertional malaise, a delayed and often prolonged worsening of symptoms triggered by even minor physical or cognitive exertion. Unlike ordinary fatigue, post-exertional malaise can last days or weeks and frequently leaves patients functionally incapacitated. Despite affecting an estimated three million people in the United States, ME/CFS has no FDA-approved treatments, and its underlying biology has remained difficult to pin down.
What makes this trial relevant to the broader mTOR story is the mechanistic hypothesis driving it. Researchers at the University of Wisconsin-Milwaukee had previously observed something unusual in ME/CFS patients: elevated levels of a protein called ATG13, which is essential for initiating autophagy. But closer inspection revealed a paradox. The ATG13 was heavily phosphorylated at a specific site, rendering it functionally inactive. The machinery required to initiate cellular cleanup was present, but locked in an off position. The upstream culprit, the protein responsible for that inactivating phosphorylation, was mTOR.
In animal models, genetic disruption of ATG13 produced severe exercise-induced fatigue, and pharmacological activation of mTOR generated similar effects, including muscle inflammation and nerve demyelination. The hypothesis that emerged was consequential: ME/CFS may represent a state of impaired autophagy driven by inappropriate mTOR activation, leaving cells unable to recover from even modest energetic stress. If true, restoring autophagy rather than managing symptoms downstream might address a root biological vulnerability of the disease.
The trial enrolled 86 individuals with ME/CFS and administered low-dose rapamycin at 6 mg once weekly, a schedule designed to modulate mTOR signaling without continuous suppression. Forty participants completed the full 90-day protocol. The results were notable on two levels: clinical and molecular.
The Clinical Picture
Clinically, participants reported broad improvements across the core features of ME/CFS. Physical activity levels increased by approximately 15% on the Bell Activity Scale, a meaningful shift in a population where even minor exertion can trigger prolonged symptom crashes. Fatigue severity declined significantly across all five domains of the Multidimensional Fatigue Inventory, encompassing general fatigue, physical exhaustion, reduced activity, diminished motivation, and mental fatigue, with the aggregate score improving at every follow-up time point. Post-exertional malaise, the disease's defining feature, showed some of the strongest and most sustained improvements, with reductions reaching the highest levels of statistical significance at multiple time points. Sleep disturbance and orthostatic intolerance also declined meaningfully over the study period.
Crucially, no significant changes were observed in metabolic, lipid, or hematologic safety markers across 90 days, reinforcing the growing evidence that intermittent low-dose rapamycin carries a meaningfully different risk profile than the continuous high-dose regimens used in transplant medicine.
What the Autophagy Data Showed
The molecular data provided the more compelling part of the story, and it is worth understanding what the two key markers actually measure before examining what happened to them.
The first is BECLIN-1, a protein that helps initiate the formation of autophagosomes, the temporary membrane structures that enclose cellular debris so it can be safely broken down and recycled. Think of BECLIN-1 as a signal that the cell's cleanup crews are being actively assembled and dispatched. Rising BECLIN-1 levels indicate that the autophagy process is being engaged more robustly.
The second is phosphorylated ATG13, which represents the opposite: a molecular roadblock. When mTOR phosphorylates ATG13 at a specific site, it inactivates the protein and prevents autophagy from getting started. High levels of this modified form indicate that the cell's recycling system is being actively suppressed before it can run.
Over the 90-day treatment period, both markers moved in the direction the hypothesis predicted. BECLIN-1 rose by roughly 40 to 50% from baseline by the second and third follow-up visits, indicating that cellular cleanup machinery was being assembled at a meaningfully higher rate. Simultaneously, phosphorylated ATG13 fell by more than two-fold, indicating that the molecular brake on autophagy was being progressively released. More recycling machinery being assembled, and fewer signals blocking it from running: the two changes together suggest that rapamycin was restoring the conditions under which cellular maintenance could actually proceed.
When the Biology Tracked the Symptoms
What made these findings particularly compelling was how closely the molecular changes tracked the clinical outcomes. Patients whose cellular cleanup systems showed the greatest restoration were the same patients reporting the largest improvements in how they felt and functioned day to day.
As BECLIN-1 levels rose, patients reported meaningful gains in physical activity tolerance, energy, and emotional well-being. As the molecular brake on autophagy released, activity tolerance improved in parallel. These were not independent observations happening to move in the same direction. The patients showing the clearest signs of restored autophagy were the ones recovering most from the symptoms that define ME/CFS.
This relationship was particularly strong among patients classified as clinical responders, who showed up to three-fold increases in BECLIN-1 alongside early and sustained reductions in phosphorylated ATG13. Partial responders showed intermediate changes. Non-responders showed little consistent shift in either autophagy marker. The biological heterogeneity of response, in other words, appeared to reflect genuine underlying differences in how much mTOR-mediated autophagy suppression was present and how readily it could be reversed, not simply noise in a complex disease population.
The Post-Infectious Signal
The alignment between cellular repair and symptom improvement was most striking in patients whose illness had followed a documented viral infection, a subgroup long suspected to represent a biologically distinct form of ME/CFS. In these patients, rapamycin produced deeper and more consistent autophagy biomarker changes alongside greater clinical improvement. BECLIN-1 rose more robustly, phosphorylated ATG13 fell by nearly three-fold by the study's end, and the improvements in quality of life, physical function, and mental health scores were the strongest in the cohort. The finding suggests that post-infectious ME/CFS may be characterized by a particularly persistent block in cellular stress-response pathways, one that mTOR inhibition is especially well positioned to release.
What This Trial Adds to the Broader Story
The study has significant limitations. It was observational rather than placebo-controlled, relied on self-reported outcomes, and experienced substantial attrition driven largely by financial barriers rather than safety concerns. These constraints mean causality cannot be established from this data alone.
But the ME/CFS trial contributes something the healthy aging trials cannot: a mechanistic stress test. ME/CFS patients represent a population in which the cellular recovery machinery appears to be specifically and persistently impaired. The fact that low-dose rapamycin, applied at the same intermittent dosing schedule used in the PEARL trial, produced measurable restoration of autophagy signaling alongside meaningful symptom relief provides some of the most direct human evidence yet that mTOR modulation can engage cellular maintenance pathways in living patients, and that doing so may translate into clinically meaningful outcomes.
For the longevity field, the implications extend beyond ME/CFS. The same autophagy impairment documented in these patients, the accumulation of cellular debris, the suppression of repair processes, the inability to recover efficiently from metabolic stress, is increasingly understood as a feature of biological aging itself. What this trial demonstrates is that these processes can be measured, modulated, and potentially corrected in humans. That is a finding with relevance well beyond any single disease.
Safety and Dosing: Reframing the Risk Profile
For decades, rapamycin's reputation in clinical medicine has been shaped by its original use as an immunosuppressant in organ transplantation. At the high continuous doses required to prevent rejection, it carries meaningful risks: metabolic disturbances, impaired wound healing, elevated infection susceptibility, and lipid abnormalities. That history has made clinicians and patients understandably cautious about its use outside of transplant medicine. The PEARL trial's safety data begin to complicate that picture in important ways.
Over 48 weeks of low-dose, intermittent use in healthy older adults, no significant changes were observed in blood glucose, liver function, or kidney markers. The metabolic harms that characterize high-dose continuous rapamycin were simply absent. This is consistent with a growing body of evidence suggesting that the risk profile of rapamycin is not fixed but highly dependent on dose and dosing schedule. A drug taken daily at transplant doses and a drug taken once weekly at a fraction of that exposure are, in a meaningful physiological sense, different interventions engaging the same pathway with very different intensity and duration.
Two additional safety observations are worth noting. First, participants in the placebo group reported a higher incidence of mouth sores than those receiving rapamycin, inverting the concern that the drug would cause this common side effect. Second, the trial coincided with the COVID-19 pandemic, providing an unplanned but informative window on immune function. Rates of reported infection were approximately 20% in the rapamycin groups compared to roughly 25% in the placebo group. While this was not a prespecified endpoint and should be interpreted cautiously, it aligns with prior evidence suggesting that intermittent mTOR inhibition may support immune resilience by limiting the chronic activation that leaves the immune system less responsive to acute challenges, precisely the mechanism explored in the Mannick immunosenescence study discussed earlier.
A Dosing Nuance With Significant Implications
Perhaps the most practically important finding to emerge from the PEARL trial's safety analysis was not about adverse effects at all, but about how much rapamycin participants were actually receiving.
The formulation used in the trial was approximately 3.5 times less bioavailable than standard oral rapamycin. This means that the nominal 10 mg weekly dose, the one producing measurable improvements in muscle mass and bone mineral content, likely corresponded to an effective systemic exposure closer to 2.9 mg of standard rapamycin. In other words, the benefits observed in the trial may have been achieved at a pharmacologically modest dose, one that sits well below what most longevity-focused rapamycin protocols currently use.
This finding carries two important implications. The first is reassuring: the mTOR pathway governing cellular maintenance and repair appears sensitive enough to respond to relatively low levels of inhibition, suggesting that meaningful biological effects do not require aggressive dosing. The second is a caution: it means that direct comparisons between the PEARL trial's dosing and other rapamycin protocols require careful attention to formulation and bioavailability, not just the number on the pill. As the field moves toward more refined and individually tailored approaches to mTOR modulation, dose optimization and pharmacokinetic monitoring will be essential tools for translating promising trial results into reliable clinical outcomes.
Natural Modulators of mTOR Signaling
Rapamycin represents one end of a spectrum. It binds directly to its molecular target, produces predictable and dose-dependent effects, and has now demonstrated measurable benefits in human clinical trials. But it is a pharmaceutical agent, and not everyone considering longevity interventions is ready to adopt a prescription drug, however favorable its intermittent low-dose profile appears to be.
At the other end of the spectrum sit the plant-derived compounds that have drawn interest from researchers for their ability to influence mTOR-related pathways through more indirect and gradual means. These phytochemicals do not switch the mTOR pathway off the way rapamycin does. They nudge it, engaging upstream regulators and competing for molecular binding sites in ways that shift the balance between growth and maintenance without fully suppressing either.
A 2026 study published in the ACS Journal of Natural Products examined this interaction systematically, describing the relationship between natural compounds and mTOR in terms of mechanistic plausibility [10]. The key finding was that certain plant-derived molecules possess structural features that allow them to physically interact with components of the mTOR signaling complex, not merely by influencing cellular metabolism generally, but by competing with adenosine triphosphate, the molecular fuel that kinases like mTOR require to function. By occupying the binding sites that ATP would otherwise fill, these compounds can reduce the intensity of mTOR's signaling activity and slow the downstream processes that drive cellular growth.
A second mechanism operates further upstream. Several of these compounds modulate the PI3K-Akt pathway, the signaling cascade that governs whether mTOR gets activated in the first place. Quieting this upstream pathway has a downstream effect: mTOR activity falls, autophagy is partially released from suppression, and the cell shifts incrementally toward the maintenance and recycling processes that chronic mTOR activation had been keeping in check.
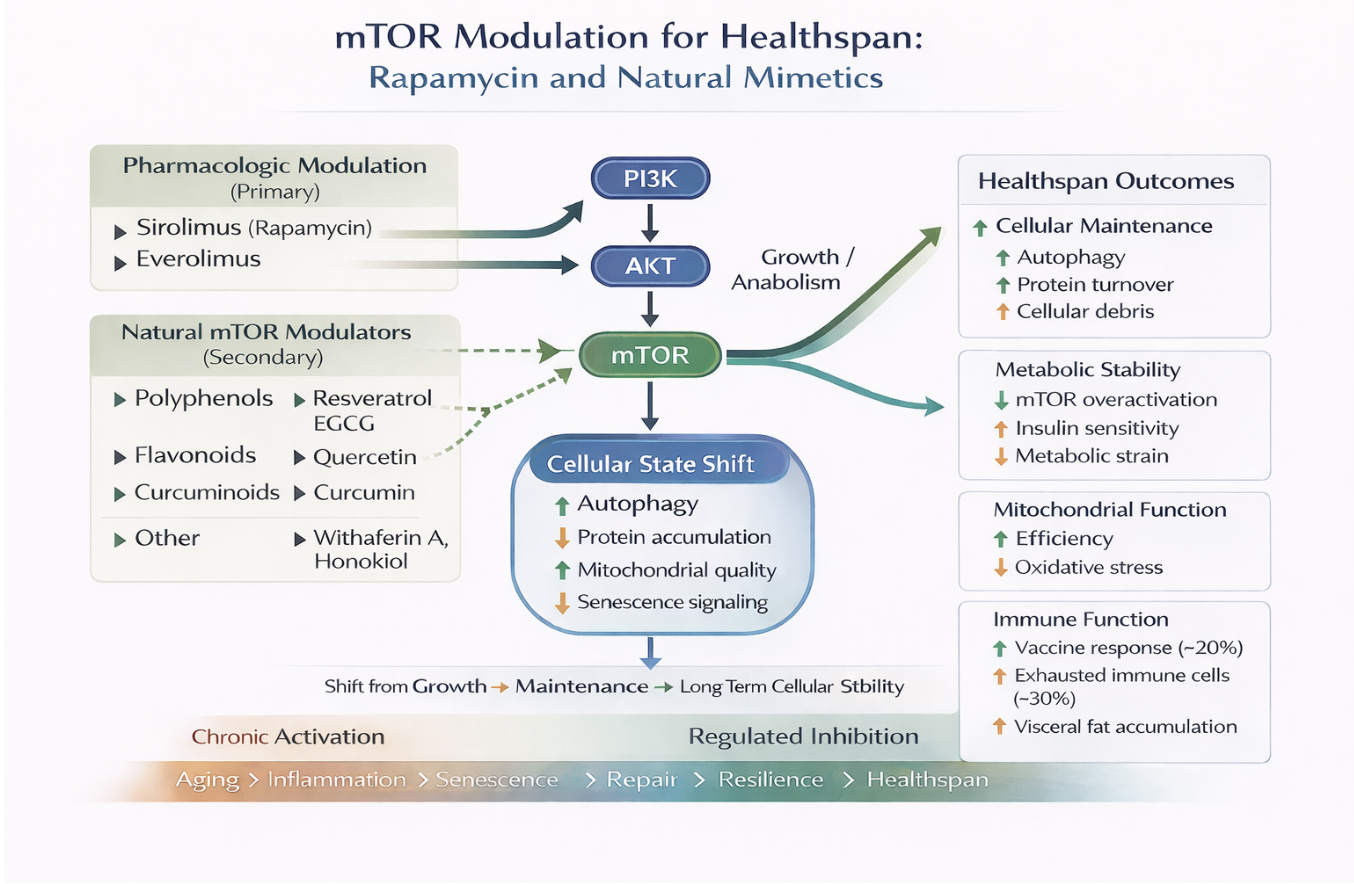
Figure 4. mTOR Modulation for Healthspan. mTOR can be regulated by pharmacological and natural interventions which also includes nutrition and lifestyle inputs.
The result is a more context-dependent and gradual form of modulation than rapamycin provides. Whether that translates into meaningful biological benefit in humans is a question the evidence will need to answer. But the mechanistic logic is coherent, and several of these compounds have been studied extensively enough to illustrate how natural mTOR modulation actually operates at the cellular level. The sections that follow examine the most thoroughly investigated of them in turn.
Resveratrol: Mimicking Energy Stress to Promote Cellular Repair
Resveratrol has a cultural backstory that precedes its science. For decades, the so-called French paradox, the observation that populations consuming moderate amounts of red wine appeared to enjoy lower rates of cardiovascular disease despite diets rich in saturated fat, prompted researchers to look closely at the compounds in grape skins and berries. Resveratrol, a polyphenolic molecule concentrated in those sources, became one of the most studied natural compounds in longevity biology. Whether the paradox itself holds up to scrutiny is a separate question. What the cellular research has revealed about resveratrol's mechanisms is more interesting than the wine story that preceded it.
At the cellular level, resveratrol operates through a principle called hormesis: the idea that mild stress, applied at the right dose, can make a biological system more resilient rather than damaging it. Rather than directly inhibiting mTOR the way rapamycin does, resveratrol activates AMPK, the cellular fuel gauge introduced earlier in this article. By activating this energy-sensing pathway, resveratrol mimics the signal of energy scarcity without actually starving the cell. The cell reads the signal, dials back mTOR activity, and begins the maintenance processes that mTOR had been suppressing, most notably autophagy [11].
The downstream evidence of this shift is visible in the molecular machinery of cellular cleanup. Autophagy activation is associated with upregulation of proteins including ATG5 and LC3-II, which are essential components of the autophagosome assembly process. Think of these proteins as the scaffolding and labeling system that allows the cell to identify, package, and deliver damaged components to the lysosome for recycling. Their upregulation signals that the cleanup process is not merely being initiated but actively progressing.
Resveratrol's effects are also dose-dependent in a way that is worth understanding clearly. At lower doses, it behaves as a cellular maintenance signal, reducing oxidative stress, supporting mitochondrial function, and promoting the protein turnover that keeps cells functioning efficiently. At higher doses, its effects shift toward something more aggressive. In environments with elevated copper levels, which tend to characterize metabolically stressed or abnormal tissues, resveratrol can generate pro-oxidant conditions that facilitate DNA damage and push abnormal cells toward programmed cell death. This biphasic behavior, beneficial at low doses and selectively destructive at high ones, is a recurring theme in hormetic compounds and one that complicates simple dosing recommendations.
A practical limitation tempers the enthusiasm for resveratrol. It is rapidly metabolized in the gut and liver, which means that a relatively small fraction of what is consumed ever reaches systemic circulation at biologically active concentrations. A related compound called pterostilbene, found in blueberries, has attracted interest as a more bioavailable alternative. Its greater lipophilicity, meaning it dissolves more readily in fats than in water, allows it to cross cell membranes more efficiently and remain stable in circulation longer, while appearing to engage similar biological pathways.
Resveratrol remains one of the more mechanistically coherent natural modulators of the mTOR pathway, operating through AMPK activation rather than direct mTOR binding. But the gap between its cellular biology and its real-world impact in humans, partly a consequence of its bioavailability constraints, is one of the themes that will recur as we examine the compounds that follow.
Curcumin and EGCG: Targeting Inflammation and Cellular Cleanup
If resveratrol works primarily by mimicking energy stress, curcumin takes a different route into the same territory. Where resveratrol activates AMPK to indirectly suppress mTOR, curcumin intervenes more directly in the signaling cascade that drives mTOR activation in the first place, while simultaneously targeting the inflammatory processes that chronic mTOR activity tends to generate.
The pathway curcumin most directly engages is the same PI3K-Akt-mTOR signaling cascade introduced earlier, the molecular chain of command that tells cells when to grow and when to hold back [12]. Rather than interrupting mTOR directly, curcumin works further upstream, interfering with several of the relay signals that would normally pass the growth instruction down the chain. Think of it like disrupting a telephone game at multiple points simultaneously: if the message gets garbled or blocked before it reaches the end of the line, the final instruction never arrives with full force.
The result is a quieter signal reaching mTOR, and a cell that consequently shifts its priorities incrementally toward maintenance rather than expansion. What makes curcumin's approach distinctive is that it engages this dampening effect through several redundant mechanisms at once, not just blocking the growth signal from being sent, but also actively promoting the molecular processes that remove those signals once they have been generated. It is less like a single switch being flipped and more like multiple hands applying pressure to the same brake.
The anti-inflammatory dimension of curcumin's activity operates in parallel and is arguably as important as its effect on mTOR signaling. One of the downstream consequences of chronic mTOR activation is the sustained production of pro-inflammatory signaling molecules, the same cytokines including IL-6 and TNF-alpha that visceral fat generates and that accumulate in the chronic low-grade inflammatory state associated with aging. Curcumin suppresses the pathway that drives their production, reducing the inflammatory background noise that both reflects and reinforces cellular aging. Through this dual action on growth signaling and inflammation, curcumin addresses two of the primary mechanisms by which persistent mTOR activity contributes to age-related cellular decline.
Preclinical research has begun to extend this picture into the domain of brain aging specifically. A 2024 study by Akansha and colleagues used a well-established animal model of accelerated aging, in which rats were administered a compound called D-galactose to induce the oxidative stress, mitochondrial dysfunction, and gene expression changes associated with biological aging. Animals that received curcumin alongside the aging-inducing treatment showed meaningfully lower oxidative stress markers and improved mitochondrial function compared to those that did not. More relevant to the broader argument of this article, curcumin supplementation was associated with upregulation of key autophagy-initiating genes, including BECLIN-1 and ULK1, the same proteins whose restoration in ME/CFS patients tracked so closely with clinical improvement. SIRT1, a protein involved in cellular stress resistance, DNA repair, and longevity signaling, was also upregulated.
These findings reinforce the biological coherence of curcumin's mechanism. Its effects are not limited to quieting growth signals upstream of mTOR. It also appears to actively engage the downstream repair and recycling processes that mTOR suppression is meant to release. Whether these effects translate into meaningful outcomes in humans, and at what doses, remains an open question that the bioavailability constraints discussed in the following section make more complicated than the preclinical data alone would suggest.
EGCG: Opening the Door to Autophagy
Epigallocatechin gallate, better known as EGCG and most familiar as the primary polyphenol in green tea, approaches the mTOR-autophagy axis from yet another direction. Rather than suppressing the signals that keep mTOR active, EGCG works by directly engaging the machinery that initiates autophagy, essentially pushing the cellular cleanup door open from the other side [13].
To understand how this works, it helps to think of autophagy as a process that requires a specific ignition event before it can begin. EGCG activates the molecular switch that triggers that ignition, prompting the cell to begin assembling the temporary membrane structures that enclose damaged proteins and dysfunctional organelles so they can be safely broken down and recycled. Without that ignition signal, the cleanup machinery sits idle even when mTOR is not actively suppressing it. EGCG provides the spark.
This matters particularly under conditions of cellular stress. When proteins misfold and accumulate inside cells, as they do during periods of metabolic strain or in aging tissue, the resulting pressure can push cells toward programmed death rather than repair. EGCG supports cellular survival under these conditions by helping keep the repair window open longer, giving stressed cells more time and biological resources to recover before the decision to eliminate them is made. Think of it as extending the grace period during which a cell can still clean house rather than being cleared away entirely.
EGCG also facilitates the removal of lipid droplets and protein aggregates, the accumulated byproducts of cellular metabolism that contribute to dysfunction when left to build up over time. This makes it particularly relevant to brain aging, where protein aggregate accumulation, including the deposits associated with neurodegeneration, represents one of the primary mechanisms of long-term cellular decline.
Together, curcumin and EGCG represent two distinct strategies for shifting cellular behavior toward maintenance. Curcumin quiets the upstream signals that keep mTOR active and inflammation elevated. EGCG directly engages the autophagy machinery that mTOR suppression is meant to release. Neither approaches the potency or predictability of rapamycin, and both face bioavailability constraints that limit how much of their cellular activity translates into systemic effect. But their mechanisms are coherent, and they point toward the possibility that a carefully chosen combination of natural modulators might engage the mTOR-autophagy axis through multiple complementary entry points simultaneously.
Resveratrol, curcumin, and EGCG represent the natural compounds with the most developed mechanistic literature and the earliest signals of human relevance. The three examined next sit in different territory. Honokiol, withaferin A, and berberine have been studied primarily in cells and animal models, with human data considerably thinner. Their value in this framework is less as established interventions than as mechanistic illustrations of how many different points of leverage the mTOR pathway actually offers: the interface between mTOR signaling and immune surveillance, the coordinated engagement of multiple upstream regulators at once, and the direct suppression of cellular senescence. That variety matters for the combination strategies discussed later in this article, which depend on having distinct entry points to work with.
Honokiol: Enhancing Immune Surveillance Through mTOR Modulation
Honokiol, a bioactive compound derived from magnolia bark, introduces a dimension of mTOR modulation that the other natural compounds examined so far have not addressed directly: the relationship between mTOR signaling and the immune system's ability to identify and clear dysfunctional cells.
To understand why this matters, it helps to know that the immune system depends on being able to distinguish healthy cells from damaged or abnormal ones. Healthy cells display molecular signals on their surface that essentially communicate to immune cells: nothing to see here, move along. Abnormal cells, including those that have accumulated the kind of damage that aging progressively generates, sometimes exploit this same signaling system to hide from immune surveillance, displaying proteins that tell the immune system to stand down even when intervention would be appropriate.
One of the key proteins involved in this evasion is called PD-L1, which functions like a molecular camouflage flag. When a cell displays high levels of PD-L1, immune cells recognize it as off-limits and leave it alone. What makes honokiol interesting is that it appears to reduce PD-L1 expression selectively in abnormal cells by modulating the mTOR signaling pathway that drives its production [14]. In practical terms, this strips away the camouflage, making dysfunctional cells more visible to the immune system without disrupting the protective signals on healthy cells.
The distinction between selective and broad immune modulation matters here. Many interventions that enhance immune activity do so indiscriminately, which carries its own risks. Honokiol's apparent selectivity, reducing immune evasion in abnormal cells while leaving the activity of healthy immune cells, including their ability to produce the signaling molecules that coordinate an effective response, largely intact, represents a more targeted form of immune support. The immune system retains its precision rather than simply being amplified.
The research on honokiol remains primarily preclinical, and the human evidence base is considerably thinner than that for rapamycin or even for resveratrol and curcumin. But its mechanism adds an important dimension to the broader picture of what mTOR modulation can accomplish: not just shifting the cell's internal balance between growth and maintenance, but influencing how the immune system interacts with cells that have drifted toward dysfunction. In the context of aging, where both mTOR dysregulation and declining immune surveillance contribute to the accumulation of damaged and senescent cells, that combination of effects is worth watching.
Withaferin A: Balancing Growth and Repair Through Dual Pathway Control
Ashwagandha has been used in Ayurvedic medicine for centuries as an adaptogen, a substance believed to help the body resist physical and psychological stress. Modern molecular biology has begun to examine the mechanisms behind that traditional reputation, and one of its primary bioactive compounds, withaferin A, has emerged as a particularly interesting candidate for mTOR-related research [15].
What distinguishes withaferin A from the other natural compounds examined in this section is the breadth of its reach across the cellular aging machinery. Rather than modulating a single upstream regulator or engaging one entry point into the mTOR pathway, it appears to influence several of the key systems that collectively determine whether a cell ages gracefully or tips toward dysfunction.
The first mechanism is familiar from earlier sections. Withaferin A activates AMPK, the cellular fuel gauge that signals energy scarcity and suppresses mTOR activity in response. By engaging this pathway, it shifts the cell's resource allocation away from growth and toward conservation and repair, mimicking at the molecular level some of the effects associated with caloric restriction without requiring an actual reduction in food intake.
The second mechanism operates in tandem. While AMPK activation suppresses mTOR from one direction, withaferin A also appears to reduce mTOR activity more directly, reinforcing the shift toward cellular maintenance through a complementary route. The combined effect of these two actions may be particularly relevant for a process called gero-conversion, the gradual transition of functional cells into a senescent state. Senescent cells, as introduced earlier in this article, are cells that have lost the ability to divide or function properly but refuse to undergo programmed death, instead lingering in tissue and secreting inflammatory signals that progressively impair the cells around them. By suppressing the growth-promoting signals that drive gero-conversion, withaferin A may help keep functional cells from crossing that threshold.
Beyond its effects on AMPK and mTOR, withaferin A influences two additional regulators that sit at the intersection of growth, stress response, and longevity. The first is IGF-1, a growth factor that, when chronically elevated, is associated with accelerated cellular aging. Think of IGF-1 as a persistent instruction to the cell to keep growing and dividing, an instruction that is useful in development but increasingly counterproductive in older tissue. Withaferin A appears to dampen this signal, reducing one of the upstream pressures that keeps mTOR chronically active. The second is SIRT1, a protein that functions as a cellular stress manager, coordinating DNA repair, autophagy activation, and resistance to oxidative damage. Enhancing SIRT1 activity effectively strengthens the cell's capacity to respond to and recover from the stresses that accumulate with age.
What withaferin A's profile suggests, more than any of the other natural compounds examined in this article, is that the mTOR-aging axis is not a single dial to be turned but a system with multiple points of leverage. Withaferin A appears to apply pressure to several of those points simultaneously, engaging AMPK, suppressing mTOR, reducing IGF-1, and enhancing SIRT1 in a coordinated fashion that no single-mechanism compound can replicate. Whether that breadth translates into greater real-world benefit remains to be established in human trials. But it raises an intriguing possibility for the combination strategies discussed later in this article: that a compound capable of modulating multiple upstream inputs simultaneously might be particularly well suited as a complement to the precision of direct mTOR inhibition with rapamycin.
Berberine: A Natural Compound With Gero-Suppressive Properties
Berberine is an isoquinoline alkaloid with a history of medicinal use spanning centuries, appearing in both Ayurvedic and traditional Chinese medicine. In recent years it has attracted serious scientific attention, not for any single therapeutic application, but for something more fundamental: evidence that it may suppress the cellular processes that drive biological aging itself.
The mechanism begins in the mitochondria. Berberine localizes within these organelles, where it interferes with the electron transport chain, the series of molecular complexes that power ATP production. This might sound like a problem, but the consequence is one that longevity researchers have been deliberately trying to induce. When mitochondrial energy production is partially impaired, the ratio of AMP to ATP inside the cell rises, and that shift activates AMPK, the cellular fuel gauge introduced earlier in this article. AMPK activation, in turn, suppresses mTOR signaling. The chain of events is essentially the same as the one triggered by caloric restriction or intense exercise, but initiated by a small molecule rather than a behavioral intervention. Notably, this mechanism closely parallels that of metformin, one of the most widely studied longevity drugs, which also targets mitochondrial complex I to activate AMPK.
A 2013 study published in the journal Aging provided one of the more direct demonstrations of berberine's gero-suppressive potential. Researchers at New York Medical College induced premature cellular senescence in human lung cells using a DNA-damaging agent, then examined whether berberine could attenuate that process. Senescence, as established earlier in this article, is the state in which a cell loses the ability to divide or function properly but refuses to undergo programmed death, instead lingering in tissue and secreting inflammatory signals that degrade the surrounding cellular environment.
The results were striking. Berberine reduced multiple hallmarks of senescence in a dose-dependent fashion. The characteristic physical changes that accompany cellular senescence, cells becoming enlarged and flattened rather than compact and functional, were substantially reduced. A cellular enzyme called beta-galactosidase, which accumulates in senescent cells and serves as one of the most reliable markers of their presence, showed reductions of up to 75% in berberine-treated cells compared to those exposed to the senescence-inducing agent alone. A protein called p21, which acts as a molecular brake on cell division and accumulates as cells enter senescence, was reduced by up to 94% at higher berberine concentrations.
Importantly, these changes were accompanied by measurable reductions in mTOR signaling activity, specifically in the phosphorylation of a downstream mTOR target that reflects how actively the pathway is driving cellular growth. The suppression of mTOR signaling and the attenuation of senescence moved together, providing direct evidence that berberine's gero-suppressive effects operate through the same pathway that rapamycin targets pharmacologically.
Berberine also reduced markers of DNA damage in the treated cells. This is significant because chronic mTOR activation and oxidative DNA damage are not independent processes. Active mTOR drives protein synthesis, which requires energy production through oxidative metabolism, which generates reactive oxygen species that can damage DNA. By quieting mTOR through AMPK activation, berberine appears to reduce the cellular energy expenditure that feeds this cycle, lowering both the growth pressure and the oxidative burden simultaneously.
The study further demonstrated that berberine's effects were apparent at concentrations considered pharmacologically relevant to what can be achieved in living organisms through oral supplementation, not just at the artificially high doses that sometimes make laboratory findings difficult to translate into practice.
What makes berberine particularly compelling in the context of this article's broader argument is not just its mechanism but its history. Most longevity interventions are recent discoveries, compounds identified through modern screening or repurposed from other medical contexts. Berberine has been used clinically for centuries across cultures that had no knowledge of mTOR, and yet the pathway it engages turns out to be one of the most central in modern aging biology. That convergence, between ancient empirical medicine and contemporary molecular science, is not proof of efficacy in humans. But it is a signal worth taking seriously, and one that justifies the rigorous human trials that are still needed to determine how much of berberine's cellular biology translates into measurable healthspan benefit.
Direct vs. Indirect Modulation of mTOR Signaling
The natural compounds examined in the preceding sections all engage the mTOR pathway, but none of them engages it the way rapamycin does. Understanding that distinction is essential for evaluating what each approach can realistically deliver, and for thinking clearly about where they might work best together.
The difference comes down to where in the biological chain of command each intervention applies its influence.
Direct Inhibition: Precision and Power of Rapamycin
Rapamycin acts at the source. It binds to an intracellular protein called FKBP12, and the resulting complex attaches directly to mTOR and inhibits its activity. There is no relay, no upstream signal to amplify, no cellular context required for the intervention to take effect. Rapamycin finds its target and switches it off with a directness that no plant-derived compound currently replicates.
This directness translates into predictability. Because rapamycin's mechanism is well characterized and its binding to mTOR is specific, its effects are dose-dependent in ways that allow researchers and clinicians to calibrate the intervention with precision. The PEARL trial demonstrated this concretely: by adjusting dose and dosing schedule, the researchers could produce measurable improvements in muscle mass and bone mineral content in older adults while maintaining a favorable safety profile. That kind of precision is the signature of a direct inhibitor operating through a defined molecular mechanism.
Indirect Modulation: Subtle but Limited Effects of Phytochemicals
Natural compounds work further upstream. Rather than binding mTOR directly, they influence the regulators that feed into it, activating AMPK to suppress mTOR from one direction, or quieting the PI3K-Akt signaling cascade that drives mTOR activation from another. The cell interprets these upstream signals and adjusts mTOR activity in response, but the adjustment is filtered through the cell's own regulatory logic. How much mTOR activity changes depends on the compound, the dose, the cellular context, and the metabolic state of the tissue in question.
This indirectness introduces two practical constraints that the evidence makes clear.
Lower Potency:
The first is potency. Natural compounds generally require concentrations substantially higher than what standard supplementation achieves in order to produce meaningful mTOR inhibition in laboratory settings. Resveratrol, for example, has been shown to suppress mTOR signaling at concentrations of approximately 20 to 50 micromolar, levels that far exceed what circulates in the bloodstream after a typical oral dose [16]. The compound can clearly influence the pathway in a controlled cellular environment. Whether it does so meaningfully in a living person taking a capsule is a different question.
Limited Bioavailability:
The second constraint is bioavailability, the fraction of what is consumed that actually reaches the cells and tissues where it is needed. Curcumin illustrates this problem acutely. Despite producing potent effects in laboratory studies, it is rapidly broken down in the gut and liver before it can accumulate to biologically active concentrations in systemic circulation. Even at oral doses as high as 12 grams, circulating levels remain low [17]. The cellular biology is compelling. The delivery problem is real.
Together, these constraints define a critical gap between mechanistic plausibility and real-world impact, between what a compound can do when applied directly to cells in a dish and what it actually does when taken as a supplement by a person going about their day. That gap does not mean natural compounds are without value. It means their value needs to be evaluated honestly, against evidence rather than against mechanism alone, and with realistic expectations about the magnitude of effect they can produce independently.
The more productive question, and the one the field is beginning to take seriously, is not whether natural compounds can replace rapamycin. It is whether they can complement it, engaging upstream regulatory points in ways that might allow effective mTOR modulation at lower pharmacological doses. That is the logic behind combination approaches, and it is where the evidence turns next.
Stronger Together? Combining Rapamycin with Natural Compounds
The distinction between direct and indirect mTOR modulation raises a practical question. If rapamycin works at the target and natural compounds work upstream, could engaging both simultaneously produce something greater than either achieves alone?
The intuition behind this idea is sound. One of the known limitations of rapamycin is that sustained mTOR inhibition can trigger a compensatory response in which cells upregulate alternative survival pathways to compensate for the suppressed growth signal. This is a biological reality of targeting any central regulatory node: the system pushes back. Natural compounds that modulate the upstream inputs feeding into mTOR may be able to reduce that compensatory pressure, allowing rapamycin to achieve more complete pathway suppression at lower doses than would otherwise be required.
A 2015 study by Alayev and colleagues provided an early proof of concept for this logic [18]. Working in a mouse model characterized by hyperactive mTOR signaling, the researchers compared three treatment conditions: rapamycin alone, resveratrol alone, and the combination of both. The hyperactive mTOR model is particularly relevant here because it mimics a state of excessive growth signaling that resembles the chronic mTOR activation implicated in cellular aging, and it allows the researchers to measure how completely each intervention suppresses that signaling.
The combination outperformed either agent alone across every measure examined. mTOR pathway suppression was greater in the combination group than with rapamycin alone. More significantly, a protein called Akt, which functions as a survival signal and is frequently upregulated as a compensatory response when mTOR is inhibited, was reduced by approximately 45% in animals receiving the combination compared to those receiving rapamycin alone. This is precisely the compensatory rebound that combination strategies are designed to prevent: resveratrol's upstream modulation of the signaling cascade appeared to reduce the biological pressure that would otherwise drive Akt upregulation, allowing rapamycin's mTOR inhibition to hold more completely.
The downstream consequences reflected this more complete suppression. Markers of cellular proliferation fell by roughly 50% in the combination group compared to either agent alone, while the rate of programmed cell death in abnormal cells increased by nearly threefold. The cell's growth machinery was more thoroughly quieted, and its quality-control systems more actively engaged, than rapamycin or resveratrol could achieve independently [18].
It is important to be precise about what this study does and does not establish. It was conducted in an animal model of hyperactive mTOR signaling and focused on molecular and cellular outcomes rather than lifespan or systemic aging measures. The findings cannot be directly translated into recommendations for healthy adults taking rapamycin alongside resveratrol. What they provide is a mechanistically coherent proof of concept: that combining a direct mTOR inhibitor with an upstream natural modulator can produce synergistic effects that neither achieves alone, and that the mechanism for that synergy, reduced compensatory survival signaling, is biologically plausible and measurable.
For the longevity field, this suggests a direction worth pursuing with greater rigor. If natural compounds can reduce the compensatory responses that limit rapamycin's effectiveness, they might allow meaningful mTOR modulation at lower pharmacological doses, with implications for both the safety profile and the long-term sustainability of rapamycin-based longevity protocols. That hypothesis remains to be tested in larger, well-controlled human studies. But the cellular logic is sound, and the question is now squarely on the research agenda.
Fasting as a Natural mTOR Inhibitor: What Human Data Are Beginning to Show
All of the natural compounds examined in the preceding sections share a common limitation: their effects on mTOR and autophagy have been demonstrated primarily in laboratory settings and animal models, with human evidence remaining limited or indirect. The fasting-mimicking diet offers something different. It is a dietary intervention with a growing body of randomized clinical trial data, and a 2025 study published in GeroScience provides the first direct human evidence linking a structured fasting-mimicking protocol to measurable changes in autophagic flux.
The biological logic connecting fasting to mTOR inhibition is among the most well-established in the field. When nutrient availability falls, insulin and IGF-1 signaling decline, AMPK is activated by the resulting energy deficit, and mTOR activity is suppressed. The cell reads the environment as one of scarcity and pivots toward conservation and repair. Autophagy, which mTOR had been keeping suppressed, is released. This is the same cascade that caloric restriction engages, and the same cascade that rapamycin triggers pharmacologically, but initiated here through the body's own nutrient-sensing machinery.
The challenge with sustained caloric restriction is adherence. Maintaining significant calorie reduction over months or years is difficult in practice and carries potential risks. The fasting-mimicking diet was designed to circumvent this problem by delivering a short, structured period of dietary stress followed by a return to normal eating. The intervention consists of five days of a carefully formulated plant-based diet that is low in calories, protein, and sugars but relatively high in unsaturated fats, followed by refeeding. The goal is not sustained deprivation but a periodic metabolic reset that triggers the biological responses associated with fasting without requiring complete food elimination.
What the Trial Found
The Espinoza and colleagues study enrolled thirty healthy adults and randomized them to one of two fasting-mimicking diet formulations, a standard version called ProLon and a modified lower-starch version called FMD2, or a control group eating normally. Blood was collected at four time points across eight days to track both metabolic and molecular responses.
The metabolic effects were rapid and consistent across both fasting-mimicking groups. By day six, body weight had declined by approximately 1.7 kilograms in both groups while remaining stable in controls. Fasting glucose fell by roughly 14 mg/dL in the ProLon group and 13 mg/dL in the FMD2 group, while rising modestly in controls. Ketone levels, a reliable indicator that the body has shifted from glucose to fat-derived energy, increased by approximately 1.0 to 1.2 millimoles per liter in both fasting groups and remained essentially unchanged in controls. Insulin resistance improved meaningfully in both fasting groups and worsened slightly in controls over the same period.
These metabolic changes establish that even a single five-day fasting-mimicking cycle can reliably induce the physiological conditions under which mTOR suppression and autophagy activation are expected to occur. The more important question was whether autophagy was actually being engaged at the cellular level.
To assess this, the researchers measured autophagic flux in circulating immune cells, using a technique that captures not simply whether autophagy is being initiated but whether it is running to completion, meaning whether damaged cellular components are being packaged, delivered to the lysosome, and actually degraded. This distinction matters because autophagosome accumulation, which is what most static autophagy measurements detect, can reflect either increased initiation or impaired clearance. Flux measurement distinguishes between the two.
The results were notable and formulation-dependent. By day six, participants following the standard ProLon diet showed accelerated autophagosomal turnover, meaning their cellular recycling machinery was processing and clearing damaged components more rapidly than at baseline. Crucially, this elevated autophagic flux persisted even after two days of normal eating, remaining elevated on day eight. The FMD2 group showed a similar trend during the fasting period, but the effect faded after refeeding.
The divergence between the two formulations is itself informative. Both diets imposed comparable caloric restriction, yet only the standard formulation produced a sustained autophagy signal. This suggests that macronutrient composition, not merely calorie reduction, shapes how deeply the fasting signal propagates into cellular repair pathways. The specific balance of protein, starch, and fat content appears to influence whether the mTOR-suppression cascade translates into lasting autophagy engagement.
The fasting-mimicking diet study occupies a distinctive position in the landscape of non-pharmacological mTOR modulation. Unlike the natural compounds examined earlier in this article, which face significant bioavailability constraints and have demonstrated mTOR inhibition primarily in laboratory conditions, the fasting-mimicking diet has now shown measurable autophagy activation in living humans under randomized controlled conditions.
The magnitude of effect is more modest than what rapamycin produces, and the study was small with a single dietary cycle. What it offers is not a claim of equivalence with pharmacological mTOR inhibition but something arguably more important for a general audience: proof of concept that structured dietary stress can engage the cellular maintenance machinery through the body's own biology, without a prescription, and that the effect can persist beyond the fasting window itself.
In the framework this article has been building, the fasting-mimicking diet represents the dietary end of the mTOR modulation spectrum. Rapamycin sits at the pharmacological end, with direct, potent, and precisely controllable effects. Natural compounds occupy the middle ground, modulating upstream regulators with lower potency and variable bioavailability. And structured fasting offers a non-pharmacological approach that engages the same nutrient-sensing pathway through the body's own response to metabolic stress.
For individuals seeking to support cellular maintenance between rapamycin doses, or for those not yet ready to consider pharmacological mTOR inhibition, periodic fasting-mimicking cycles may represent one of the more biologically coherent dietary strategies currently supported by human data. The field is still early, and larger and longer trials will be needed to establish how much autophagic benefit accumulates across repeated cycles. But the direction of the evidence is clear: the growth-repair switch that chronic abundance leaves stuck in the wrong position can be periodically reset through the body's own nutrient-sensing machinery. The cellular recycling system can be reactivated, the senescent burden can be slowed, and the maintenance programs that aging progressively suppresses can be re-engaged, through a biological mechanism as old as scarcity itself.
What We Still Don't Know: Gaps in the Evidence
The evidence reviewed in this article makes a compelling case for mTOR as a central target in aging biology. Rapamycin extends lifespan across an extraordinary range of organisms. Low-dose intermittent use improves immune function and body composition in human clinical trials. The ME/CFS data provide rare human evidence that restoring autophagy through mTOR inhibition can produce meaningful clinical benefit. Natural compounds engage overlapping pathways through mechanisms that are biologically coherent, and preclinical data suggest they may complement direct mTOR inhibition in ways that enhance its effectiveness.
But intellectual honesty requires stating clearly what the evidence does not yet establish.
The most fundamental open question is whether rapamycin extends human lifespan. The animal data are striking, including a landmark 2009 study by Harrison and colleagues reporting that rapamycin extended lifespan in genetically diverse mice by approximately 9 to 14%, even when treatment was initiated late in life [19]. That finding was significant precisely because it demonstrated that meaningful lifespan extension was achievable even after a substantial portion of the animals' lives had elapsed, suggesting the intervention's benefits are not limited to early biological windows. But mice are not humans. The gap between lifespan extension in a rodent and the same outcome in a person is one that decades of biomedical research have repeatedly demonstrated to be wide, and sometimes uncrossable. Whether the cellular benefits observed in human trials, improved immune function, preserved muscle mass, reduced visceral fat accumulation, translate into longer lives remains unknown.
For natural compounds, the evidentiary gap is wider still. Preclinical studies consistently show that resveratrol, curcumin, EGCG, berberine, and related phytochemicals influence mTOR-related pathways in cell cultures and animal models. The mechanisms are coherent. The directions of effect are consistent with what longevity researchers want to produce. But robust, reproducible evidence that any of these compounds meaningfully influences established biomarkers of aging in humans, through mTOR-related mechanisms specifically, remains limited. The distance between a cell culture result and a clinical outcome is not merely technical. It encompasses bioavailability, pharmacokinetics, tissue distribution, individual metabolic variation, and the sheer complexity of a living human organism across decades of exposure.
Dosing is a related challenge that applies to both rapamycin and natural compounds, though in different ways. For rapamycin, the PEARL trial's bioavailability finding, that the effective dose may have been substantially lower than the nominal one due to formulation differences, underscores how much precision is still needed in translating dosing protocols across different preparations and populations. For natural compounds, the problem is more fundamental: there are currently no standardized, evidence-based guidelines for achieving consistent mTOR modulation through supplementation. Variability in absorption, gut metabolism, and tissue penetration means that two people taking the same dose of curcumin or resveratrol may achieve very different circulating concentrations, making it difficult to study these compounds rigorously or to interpret results across studies.
These gaps do not invalidate the field. They define its frontier.
Conclusion: The Healthspan Perspective
The pursuit of longevity was, for most of human history, the domain of myth. The fountain of youth, the philosopher's stone, the elixir of immortality: the language of longevity was the language of fantasy precisely because there was no biological framework capable of supporting anything more rigorous. That has changed. The question is no longer whether aging can be influenced at the molecular level. The evidence reviewed in this article makes clear that it can. The question now is how precisely, how safely, and how durably that influence can be applied across a human lifespan.
mTOR modulation sits at the center of that question in a way that few other targets in longevity research can claim. It is mechanistically central, reproducibly effective across model organisms, and increasingly supported by human data that point in the right direction even if they do not yet close the case. The human trials reviewed here do not prove that rapamycin extends lifespan. They establish something more modest but still consequential: that low-dose intermittent mTOR inhibition can measurably engage the cellular repair machinery in living people, and that the downstream effects on immune function, body composition, and symptom burden are in the direction the biology predicts. Natural compounds and structured fasting occupy different positions on the same spectrum, trading potency for accessibility and direct binding for upstream modulation. The direction of travel is becoming clearer: intermittent dosing, biomarker-driven trials, combination strategies that pair pharmacological precision with complementary upstream modulators, and improved delivery systems that might finally close the gap between the cellular biology of natural compounds and their real-world impact.
The biology of mTOR is a story about a switch that evolution calibrated for a world of scarcity and intermittent stress, and that chronic abundance has left chronically stuck in the wrong position. Rapamycin, caloric restriction, exercise, fasting, and the natural compounds reviewed in this article all push that switch in the same direction, toward maintenance, repair, and the kind of cellular resilience that healthy aging depends on. The question the field is now equipped to answer, for the first time with the tools and trial designs required to answer it properly, is how far that switch can be moved in humans, and what difference it makes to the decades of life that follow.
- Nardella, C., Carracedo, A., Alimonti, A., Hobbs, R. M., Clohessy, J. G., Chen, Z., Egia, A., Fornari, A., Fiorentino, M., Loda, M., Kozma, S. C., Thomas, G., Cordon-Cardo, C., & Pandolfi, P. P. (2009). Differential requirement of mTOR in postmitotic tissues and tumorigenesis. Science signaling, 2(55), ra2. https://doi.org/10.1126/scisignal.2000189
- Kim, Y. C., & Guan, K. L. (2015). mTOR: a pharmacologic target for autophagy regulation. The Journal of clinical investigation, 125(1), 25–32. https://doi.org/10.1172/JCI73939
- Walters, H. E., & Cox, L. S. (2018). mTORC Inhibitors as Broad-Spectrum Therapeutics for Age-Related Diseases. International journal of molecular sciences, 19(8), 2325. https://doi.org/10.3390/ijms19082325
- Johnson, S. C., Rabinovitch, P. S., & Kaeberlein, M. (2017). mTOR is a key modulator of ageing and age-related disease. Nature, 493(7432), 338–345. https://pmc.ncbi.nlm.nih.gov/articles/PMC5723685/
- Blagosklonny M. V. (2019). Rapamycin for longevity: opinion article. Aging, 11(19), 8048–8067. https://doi.org/10.18632/aging.102355
- Mannick, J. B., Del Giudice, G., Lattanzi, M., Valiante, N. M., Praestgaard, J., Huang, B., Lonetto, M. A., Maecker, H. T., Kovarik, J., Carson, S., Glass, D. J., & Klickstein, L. B. (2014). mTOR inhibition improves immune function in the elderly. Science translational medicine, 6(268), 268ra179. https://doi.org/10.1126/scitranslmed.3009892
- Girish Harinath, Lee, V., Nyquist, A., Moel, M., Morgan, S. L., Isman, A., & Sajad Zalzala. (2024). Safety and efficacy of rapamycin on healthspan metrics after one year: PEARL Trial Results. MedRxiv (Cold Spring Harbor Laboratory). https://doi.org/10.1101/2024.08.21.24312372
- LaFountain, R. (2025, December 29). Top 10 longevity & anti-aging breakthroughs of 2025: What the year taught us about extending human healthspan. Healthspan. https://www.gethealthspan.com/research/article/top-ten-longevity-anti-aging-breakthroughs-of-2025
- LaFountain, R., & Tawfik, D. (2026, February 7). Rapamycin dosing for longevity: What emerging human research reveals about how dose and timing shape autophagy without compromising metabolic health. Healthspan. https://www.gethealthspan.com/research/article/rapamycin-dosing-for-longevity
- Hu, J., Guo, Y., Ren, L., Zhang, H., Qin, X., Yin, F., & Liu, X. (2026). Natural products targeting the PI3K/Akt/mTOR-mediated autophagy pathway in cancer therapy: Recent advances and clinical perspectives. Journal of Natural Products, 89(2), 379–397. https://doi.org/10.1021/acs.jnatprod.5c01372
- Salehi, B., Mishra, A. P., Nigam, M., Sener, B., Kilic, M., Sharifi-Rad, M., Fokou, P. V. T., Martins, N., & Sharifi-Rad, J. (2018). Resveratrol: A Double-Edged Sword in Health Benefits. Biomedicines, 6(3), 91. https://doi.org/10.3390/biomedicines6030091
- Menon, V. P., & Sudheer, A. R. (2007). Antioxidant and anti-inflammatory properties of curcumin. Advances in experimental medicine and biology, 595, 105–125. https://doi.org/10.1007/978-0-387-46401-5_3
- Holczer, M., Besze, B., Zámbó, V., Csala, M., Bánhegyi, G., & Kapuy, O. (2018). Epigallocatechin-3-Gallate (EGCG) Promotes Autophagy-Dependent Survival via Influencing the Balance of mTOR-AMPK Pathways upon Endoplasmic Reticulum Stress. Oxidative medicine and cellular longevity, 2018, 6721530. https://doi.org/10.1155/2018/6721530
- Crane, C., Panner, A., Pieper, R. O., Arbiser, J., & Parsa, A. T. (2009). Honokiol-mediated inhibition of PI3K/mTOR pathway: a potential strategy to overcome immunoresistance in glioma, breast, and prostate carcinoma without impacting T cell function. Journal of immunotherapy (Hagerstown, Md. : 1997), 32(6), 585–592. https://doi.org/10.1097/CJI.0b013e3181a8efe6
- Aliper, A., Jellen, L., Cortese, F., Artemov, A., Karpinsky-Semper, D., Moskalev, A., Swick, A. G., & Zhavoronkov, A. (2017). Towards natural mimetics of metformin and rapamycin. Aging, 9(11), 2245–2268. https://doi.org/10.18632/aging.101319
- Leontieva, O. V., Paszkiewicz, G., Demidenko, Z. N., & Blagosklonny, M. V. (2013). Resveratrol potentiates rapamycin to prevent hyperinsulinemia and obesity in male mice on high fat diet. Cell death & disease, 4(1), e472. https://doi.org/10.1038/cddis.2012.202
- Anand, P., Kunnumakkara, A. B., Newman, R. A., & Aggarwal, B. B. (2007). Bioavailability of curcumin: problems and promises. Molecular pharmaceutics, 4(6), 807–818. https://doi.org/10.1021/mp700113r
- Alayev A, Salamon RS, Sun Y, et al. Effects of combining rapamycin and resveratrol on apoptosis and growth of TSC2‑deficient xenograft tumors. American Journal of Respiratory Cell and Molecular Biology. 2015;53(5):637‑646. DOI:10.1165/rcmb.2015‑0022OC. https://pmc.ncbi.nlm.nih.gov/articles/PMC4742955/
- Harrison DE, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460(7253):392–395. https://pubmed.ncbi.nlm.nih.gov/19587680/
Related studies